W diagnostyce choroby Wilsona dużą rolę odgrywa badanie genetyczne, które służy identyfikacji mutacji w genie ATP7B.14 Istotne jest także potwierdzenie obecności charakterystycznego dla tej choroby pierścienia Kaysera-Fleischera, obserwowanego u większości pacjentów z zaburzeniami neurologicznymi.15 Prawie u wszystkich chorych z objawami neurologicznymi MR głowy pokazuje nieprawidłowości.18 Zmiany występują zazwyczaj w jądrach podstawy: w skorupach, jądrach ogoniastych, wzgórzach, śródmózgowiu, moście (ryc. 3). Przeważnie obserwuje się zmiany hiperintensywne, rzadziej hipointensywne w sekwencji T2. Często występuje zanik mózgu i móżdżku.18
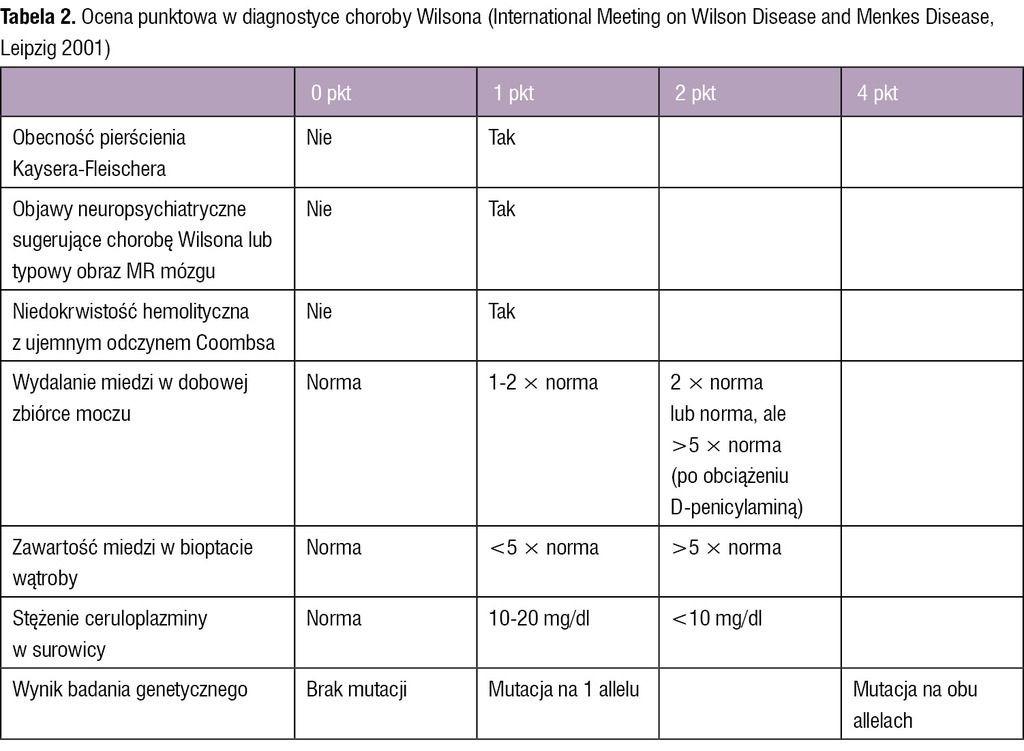
Tabela 2. Ocena punktowa w diagnostyce choroby Wilsona (International Meeting on Wilson Disease and Menkes Disease, Leipzig 2001)
Obecnie w diagnostyce choroby Wilsona zaleca się stosowanie skali oceny punktowej opracowanej w 2001 r. podczas International Meeting on Wilson Disease and Menkes Disease.19 Uzyskanie 4 lub więcej punktów w tej skali upoważnia do rozpoznania choroby Wilsona (tab. 2).
Leczenie i jego monitorowanie
Bardzo ważne jest wczesne rozpoznanie choroby i włączenie leczenia przeciwmiedziowego. Jeśli nie podejmie się terapii, postępuje uszkodzenie wątroby i narastają objawy neurologiczne. Zwykle kończy się to zgonem. Celem leczenia jest usunięcie nadmiaru miedzi z organizmu i zapobieganie jej reakumulacji. Wczesne rozpoznanie oraz regularne przyjmowanie leków przeciwmiedziowych przez całe życie może poprawić stan kliniczny lub nawet doprowadzić do całkowitego wycofania objawów klinicznych. W przypadkach z zaawansowanymi zmianami w mózgu i/lub z szybko postępującą niewydolnością wątroby leczenie może być nieskuteczne.
W Polsce w terapii choroby Wilsona stosowane są dwa leki: D-penicylamina oraz cynk. D-penicylamina jest lekiem chelatującym miedź i powoduje wydalanie jej z moczem.13,15,20,21 Lek podaje się w dawce 1,0-1,5 g/24 h, zwykle w 3 dawkach podzielonych.14 Aby zapewnić jak najlepsze wchłanianie, D-penicylaminę należy przyjmować 1 h przed posiłkiem lub 2 h po posiłku.13 Wprowadza się ją bardzo powoli (1/2 tabletki co kilka dni), ponieważ istnieje ryzyko pogorszenia stanu neurologicznego (10-50% pacjentów). Prawidłowość leczenia ocenia się, monitorując parametry metabolizmu miedzi, szczególnie stężenie wydalania miedzi w dobowej zbiórce moczu, które w przypadku leków chelatujących powinno być zwiększone.13
Drugim lekiem dostępnym w Polsce do leczenia choroby Wilsona jest cynk, który zapobiega wchłanianiu miedzi z przewodu pokarmowego. Obecnie sole cynku są zalecane głównie u pacjentów presymptomatycznych lub w terapii podtrzymującej,14 jednak według badań prowadzonych przez II Klinikę Neurologiczną skuteczność soli cynku i D-penicylaminy w terapii początkowej jest porównywalna.20,21 Proponowane dawki cynku w leczeniu choroby Wilsona wynoszą od 150 do 250 mg/24 h, podawane w 3 dawkach.13-15
Monitorowanie leczenia farmakologicznego opiera się na regularnej ocenie neurologicznej pacjentów oraz ocenie parametrów funkcji wątroby.15 W przypadku leczenia D-penicylaminą wydalanie miedzi w dobowej zbiórce moczu powinno mieścić się w zakresie 200-500 µg, a w przypadku leczenia solami cynku, które ograniczają wchłanianie miedzi z przewodu pokarmowego – wynosić mniej niż 100 µg/24 h.13-15
Przeszczepienie wątroby jest postępowaniem zarezerwowanym dla osób z chorobą Wilsona rozwijających piorunującą niewydolność wątroby oraz dla pacjentów z ciężką zdekompensowaną marskością wątroby, niereagującą na leczenie lekami chelatującymi.22
Badania przesiewowe krewnych pacjentów
Ponieważ choroba Wilsona jest dziedziczona autosomalnie recesywnie, najwyższe ryzyko zachorowania (25%) dotyczy rodzeństwa pacjenta. Teoretycznie ryzyko u potomstwa chorych jest stosunkowo niskie:13 wśród dzieci pacjentów II Kliniki Neurologicznej choroba Wilsona została stwierdzona u ok. 4%.23 Często jednak ustalenie rozpoznania u krewnych bywa trudne, ponieważ u chorych bez objawów wyniki badań metabolizmu są w granicach normy lub tylko nieznacznie zaburzone. Niewielkie zmiany w metabolizmie miedzi można stwierdzić również u heterozygot (osób z mutacją w jednym allelu genu ATP7B). Pomocne jest badanie genetyczne, zwłaszcza w diagnostyce choroby u rodzeństwa pacjenta ze znaną mutacją. Zarówno wytyczne europejskie, jak i amerykańskie zalecają badanie krewnych pierwszego stopnia.14,15
Podsumowanie
NBIA to grupa chorób z różnorodnymi zaburzeniami neurologicznymi (ruchy mimowolne, parkinsonizm, ataksje, zaburzenia poznawcze) oraz hipodensyjnymi zmianami w obrębie jąder podstawy w MR głowy w sekwencji T2. Większość zespołów chorobowych rozpoczyna się w dzieciństwie, ale niektóre również w późniejszym wieku. Do potwierdzenia rozpoznania służy badanie genetyczne. W większości zespołów NBIA leczenie jest objawowe.
Rzadki zespół chorobowy uwarunkowany genetycznie (mutacja w genie SLC30A10), związany z gromadzeniem się manganu w jądrach podstawy, cechuje się uszkodzeniem wątroby oraz obecnością objawów neurologicznych (uogólniona dystonia). W MR głowy w sekwencji T1 obserwuje się hiperintensywne zmiany zlokalizowane w jądrach podstawy i móżdżku. Leczenie opiera się na preparatach chelatujących mangan i żelazo, które u większości pacjentów są skuteczne.
W chorobie Wilsona, polegającej na zaburzeniu metabolizmu miedzi, najczęściej występują uszkodzenie wątroby oraz objawy neurologiczne (zaburzenia mowy, drżenie, zaburzenia chodu, dystonia) i psychiatryczne. Rozpoznanie opiera się na kombinacji objawów klinicznych i wyników badań laboratoryjnych, stwierdzeniu obecności pierścienia Kaysera-Fleischera oraz identyfikacji mutacji w genie ATP7B. MR pokazuje zmiany zazwyczaj w jądrach podstawy: w skorupach, jądrach ogoniastych, wzgórzach, śródmózgowiu, moście. Chorobę Wilsona można skutecznie leczyć lekami przeciwmiedziowymi.
Wiedza o patogenezie i diagnostyce genetycznie uwarunkowanych chorób OUN przebiegających z gromadzeniem metali jest coraz większa, co najprawdopodobniej przełoży się w przyszłości na lepsze możliwości terapeutyczne tych jednostek chorobowych.