Choroby genetyczne OUN przebiegające z gromadzeniem metali: żelaza, manganu i miedzi – schematy diagnostyczno-lecznicze
dr n. med. Karolina Dzieżyc1
dr hab. n. med. Tomasz Litwin1
prof. dr hab. n. med. Anna Członkowska1,2

dr n. med. Karolina Dzieżyc

dr hab. n. med. Tomasz Litwin

prof. dr hab. n. med. Anna Członkowska
Gromadzenie się metali w ośrodkowym układzie nerwowym prowadzi do ciężkich i często nieodwracalnych uszkodzeń. Toksyczny wpływ metali na ten układ może wynikać z egzogennego zatrucia związkami zawierającymi metale (np. efedron), jak również mieć podłoże genetyczne. Najlepiej poznanymi genetycznymi chorobami neurologicznymi są zespoły spowodowane gromadzeniem się żelaza, manganu i miedzi.
Wprowadzenie
W artykule omówiono trzy grupy chorób. Pierwszą jest neurodegeneracja z gromadzeniem żelaza (NBIA – neurodegeneration with brain iron accumulation), obejmująca jednostki chorobowe z różnorodnymi zaburzeniami neurologicznymi. Do grupy drugiej należy zespół chorobowy związany z gromadzeniem manganu, charakteryzujący się uszkodzeniem wątroby, policytemią oraz uogólnioną dystonią. Wreszcie choroba Wilsona, związana z zaburzeniem metabolizmu miedzi, cechuje się uszkodzeniem wątroby oraz objawami neuropsychiatrycznymi.
Neurodegeneracja z gromadzeniem żelaza
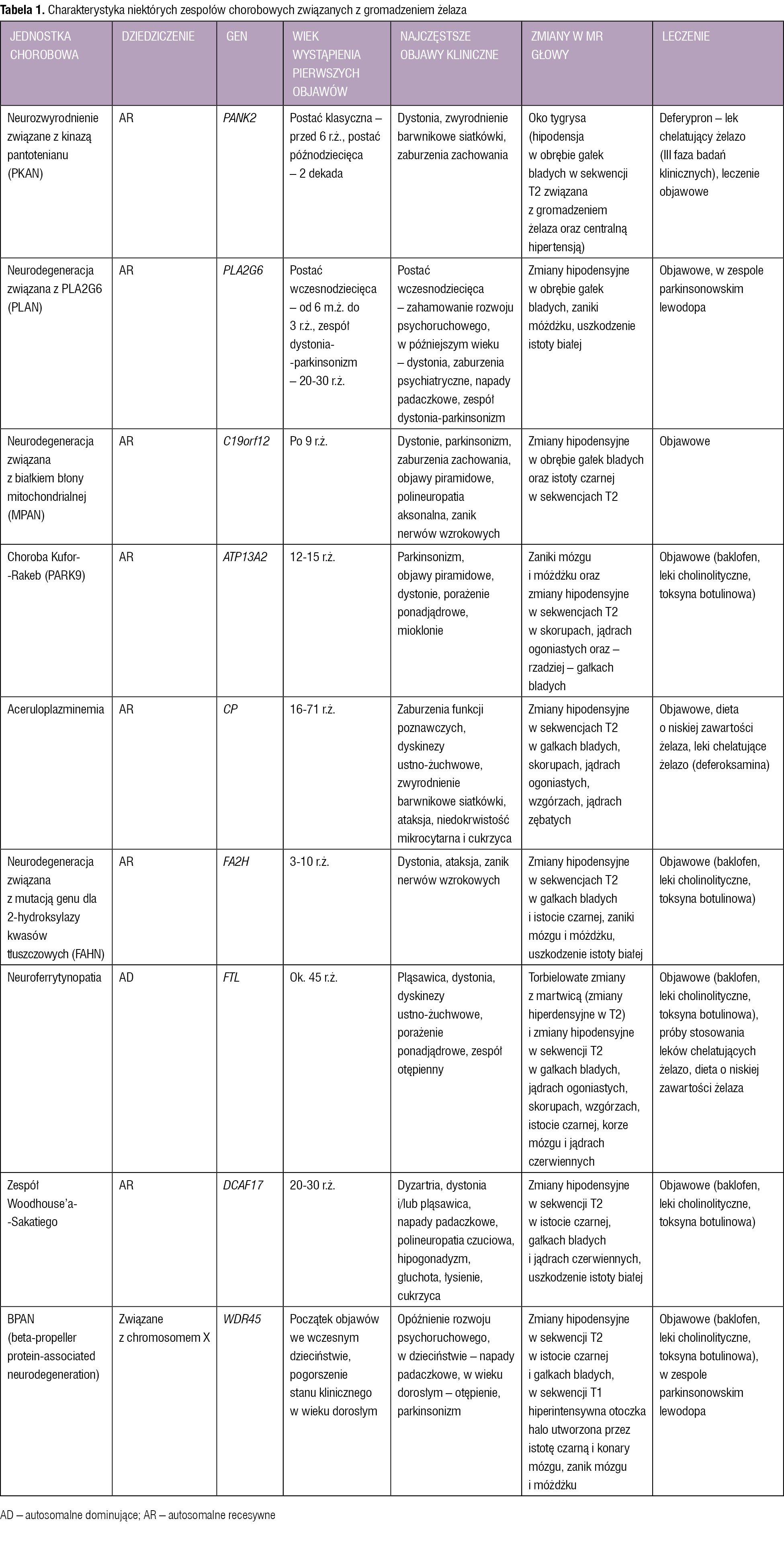
Tabela 1. Charakterystyka niektórych zespołów chorobowych związanych z gromadzeniem żelaza
NBIA to grupa chorób polegających na odkładaniu się żelaza głównie w jądrach podstawy. W NBIA występują różnorodne objawy neurologiczne, a w przypadku niektórych zespołów – także ogólnoustrojowe.1-5 Pierwsze objawy choroby mogą pojawić się w okresie od dzieciństwa do 6 dekady życia.1-5 W diagnostyce NBIA istotny jest rezonans magnetyczny (MR – magnetic resonance) głowy, w którym w sekwencji T2 stwierdza się hipodensyjne zmiany w jądrach podkorowych związane z obecnością żelaza. Dużą rolę w potwierdzeniu rozpoznania konkretnego zespołu odgrywa badanie genetyczne. W aceruloplazminemii oraz neuroferrytynopatii pierwotnie dochodzi do zaburzeń metabolizmu żelaza. W pozostałych zespołach patogeneza jest niejasna – związek z żelazem i uszkodzeniami albo nie został wytłumaczony, albo nieznany jest defekt genetyczny, a stwierdza się odkładanie tego metalu.1-5
Poniżej omówiono najczęściej występujące zespoły NBIA. Pozostałe, bardzo rzadkie jednostki chorobowe związane z gromadzeniem żelaza, m.in. chorobę Kufor-Rakeb, aceruloplazminemię, neurodegenerację związaną z mutacją genu dla 2-hydroksylazy kwasów tłuszczowych (FAHN – fatty acid hydroxylase-associated neurodegeneration), neuroferrytynopatię i BPAN (beta-propeller protein-associated neurodegeneration), przedstawiono w tabeli 1.1-8
Neurozwyrodnienie związane z kinazą pantotenianu
Neurozwyrodnienie związane z kinazą pantotenianu (PKAN – pantothenate kinase-associated neurodegeneration) to najczęściej występujący zespół (1-3/1 mln przypadków).1-4 Jego przyczyną jest mutacja w genie kodującym kinazę pantotenianu 2, która bierze udział w biosyntezie koenzymu A, niezbędnego do metabolizmu kwasów tłuszczowych. Skutkuje to zaburzeniami procesów β-oksydacji kwasów tłuszczowych i odkładaniem się L-cysteiny chelatującej żelazo.1-4
W postaci klasycznej PKAN pierwsze objawy występują przed 6 r.ż. Dominuje sztywność mięśniowa, stopniowo rozwija się uogólniona dystonia. Ponadto stwierdza się zwyrodnienie barwnikowe siatkówki oraz obecność akantocytów w rozmazie krwi. W postaci późnodziecięcej objawy pojawiają się w 2 dekadzie życia i są mniej nasilone niż w postaci klasycznej. Często występują zaburzenia zachowania. Choroba o początku objawów w wieku dorosłym cechuje się przedłużonym przebiegiem.1-4
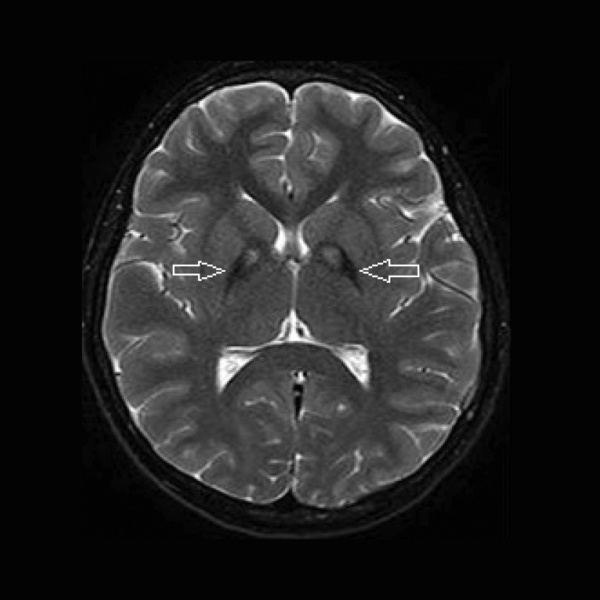
Rycina 1. MR głowy, obrazy T2-zależne. Objaw oka tygrysa u pacjenta z PKAN (zmiany hipodensyjne w obrębie gałek bladych związane z gromadzeniem się żelaza oraz centralna hiperdensja)
Charakterystyczne dla PKAN są zmiany w MR głowy. Stwierdzenie objawu oka tygrysa (hipodensja w obrębie gałek bladych w sekwencji T2 związana z gromadzeniem żelaza oraz centralną hipertensją) może być bardzo pomocne w diagnostyce choroby (ryc. 1). Rozmaz krwi obwodowej wykazuje obecność akantocytów. Istotną rolę w diagnostyce odgrywa także badanie genetyczne, które może potwierdzić rozpoznanie.1-4
W leczeniu choroby wykorzystuje się deferypron, lek chelatujący żelazo. Wyniki terapii nie zostały w pełni potwierdzone badaniami klinicznymi – dostępne są tylko obserwacje przypadków.9,10 W przypadku PKAN stosuje się obecnie leczenie objawowe. Chorym podaje się lewodopę, toksynę botulinową, baklofen i benzodiazepiny. Podejmowane są również próby leczenia głęboką stymulacją mózgu (DBS – deep brain stimulation), z różnymi efektami.1-4
Neurodegeneracja związana z PLA2G6
Neurodegeneracja związana z PLA2G6 (PLAN – PLA2G6-associated neurodegeneration) to drugi co do częstości występowania zespół NBIA. Jego przyczyną jest mutacja w genie kodującym białko PLA2G6, znajdującym się na chromosomie 22. PLA2G6 to fosfolipaza zależna od wapnia, zaangażowana w metabolizm wolnych kwasów tłuszczowych.1-4 W PLAN dochodzi do zaburzenia przepuszczalności błon komórkowych.