IV edycja Kongresu HPV już 12-13 czerwca! Poznaj najnowsze trendy w profilaktyce, diagnostyce i leczeniu raka szyjki macicy i innych schorzeń związanych z HPV | Sprawdź >
Fizjologiczne działanie hormonu wzrostu
Rola hormonu wzrostu (GH) zależy od okresu rozwojowego. GH wpływa na procesy wzrastania, powodując wzrost chrząstki przynasadowej, a za jej pośrednictwem na wzrost kości długich, pozytywnie działa także na procesy mineralizacji tkanki kostnej. Anaboliczna funkcja GH widoczna jest w układzie mięśniowym (wzrost proliferacji i różnicowania miocytów), sercowo-naczyniowym (zwiększanie liczby włókien mięśniowych lewej komory serca, wzrost objętości lewej komory serca, frakcji wyrzutowej i spadek obwodowego oporu naczyniowego), w nerkach (wzrost GFR, zwiększenie aktywności pompy sodowej, resorpcji fosforanów i syntezy kalcytriolu). Korzystny wpływ GH oś podwzgórze-przysadka-gonady jest widoczny na wszystkich jej poziomach. Działanie metaboliczne GH jest silnie lipolityczne, powodując w rezultacie zmniejszenie objętości tłuszczowej masy ciała i poprawiając profil lipidowy. U dzieci z wrodzoną ciężką postacią SNP leczenie substytucyjne rhGH zapobiega hipoglikemii przez zmniejszenie transportu glukozy do tkanek i utleniania glukozy oraz nasilenia glukoneogenezy.1,2,7
Działanie GH na tkanki docelowe przebiega w mechanizmie bezpośrednim oraz pośrednim, przez nasilenie syntezy i uwalniania insulinopodobnych czynników wzrostu (IGF), przede wszystkim IGF-1, działającego auto- i parakrynnie. IGF-1 w zdecydowanej większości krąży we krwi w postaci kompleksu związanego z białkami wiążącymi (IGF binding proteins, IGFBP), szczególnie w postaci IGFBP3. GH pobudza także wydzielanie czynników wzrostu: fibroblastów (fibroblast growth factor, FGF), nabłonka (epidermal growth factor, EGF) oraz proliferację komórek β trzustki.1,2
Leczenie niskorosłych dzieci z somatotropinową niedoczynnością przysadki
SNP spowodowana jest niedostatecznym wydzielaniem GH, co w okresie rozwojowym przejawia się upośledzeniem tempa wzrastania i znaczącym niedoborem wzrostu. Wyróżniamy izolowaną somatotropinową lub wielohormonalną niedoczynność przysadki (WNP). Oprócz niedoboru wzrostu WNP towarzyszą objawy niedoczynności innych narządów dokrewnych: wtórna niedoczynność tarczycy, nadnerczy i gonad. Niedoczynność przysadki może być uwarunkowana pierwotnie lub wtórnie.
Wrodzona postać SNP/WNP występuje z częstością 1/3500-1/10 000 urodzeń. Wrodzona SNP może być spowodowana czynnikami genetycznymi, strukturalnymi zmianami okolicy podwzgórzowo-przysadkowej lub urazem okołoporodowym. Do poznanych mechanizmów genetycznych odpowiedzialnych za niedobór GH należą mutacje genów receptora hormonu uwalniającego GH (GHRH-R) i genów GH oraz czynników transkrypcyjnych odpowiedzialnych za różnicowanie komórek przedniego płata przysadki lub rozwój przysadki (HESX1, LHX3, LHX4, SOX3, GLI2, PROP1, PIT1). W tym ostatnim przypadku mamy najczęściej do czynienia z WNP, a rodzaj zaburzeń hormonalnych wskazuje na rodzaj defektu genetycznego. Wrodzone zmiany strukturalne są zwykle związane z wrodzonymi zaburzeniami linii środkowej ciała (dysplazja przegrodowo-wzrokowa, septo-optic dysplasia, SOD).1
Wtórna postać SNP/WNP występuje u ponad 1/3 chorych i spowodowana jest chorobami rozrostowymi, najczęściej ośrodkowego układu nerwowego (OUN), oraz powikłaniami ich leczenia zarówno operacyjnego, jak i radio- czy chemioterapii. Do pozostałych rzadszych przyczyn należą m.in. urazy, stany zapalne i procesy autoimmunologiczne w obrębie OUN.1,7
Głównym objawem niedoboru GH jest zmniejszenie tempa wzrastania dziecka. W przypadku wrodzonej postaci SNP objawami sugerującym możliwość tej choroby w okresie noworodkowym i niemowlęcym mogą być wady linii pośrodkowej ciała, hipoglikemia często z hiperbilirubinemią. Niedobór GH, oprócz zwolnienia tempa wzrastania, prowadzi do wielu innych zaburzeń, takich jak: nieprawidłowa gospodarka lipidowa, miażdżyca naczyń krwionośnych, upośledzenie czynności lewej komory serca, otyłość, osteoporoza.
Preparaty rhGH wpływają zarówno na promowanie procesów wzrastania i mineralizacji kości, jak również na pozostałe procesy metaboliczne, wyrównując towarzyszące SNP nieprawidłowości (hipercholestrolemia z dyslipidemią, tendencja do hipoglikemii, zaburzenia mineralizacji kości).1,7,8
Diagnostyka SNP w okresie rozwojowym polega obecnie na wykazaniu obniżonych wyrzutów GH: w teście nocnego spontanicznego wydzielania GH (traktowanym jako test przesiewowy) oraz w dwóch testach stymulacyjnych (<10 ng/ml). Na podstawie wieloletniej obserwacji oraz danych z piśmiennictwa wydaje się celowe rozważenie rezygnacji z testu nocnego jako nieidentyfikującego niektórych chorych z SNP, słabo korelującego z wynikami testów stymulacyjnych oraz kłopotliwego do wykonania w warunkach szpitalnych. Testy dynamiczne oparte są na pobudzaniu mechanizmów zwiększających wydzielanie GH w odpowiedzi na stymulację środkiem farmakologicznym (insulina, glukagon, arginina, klonidyna, L-dopa). Na podstawie wyrzutu GH w testach stymulacyjnych można wyróżnić całkowity oraz częściowy niedobór GH (maksymalne stężenie GH odpowiednio <5 i 5-10 ng/ml). Pomocna jest także ocena stężenia IGF-1, które zazwyczaj jest znacząco zmniejszone. Badanie to jest także stosowane w trakcie monitorowania terapii rhGH.1,8-10
W każdym przypadku konieczne jest wykonanie badania obrazowego mózgu, z oceną okolicy podwzgórza i przysadki (badanie MR ze środkiem kontrastowym), w celu wykluczenia anatomicznych nieprawidłowości w obrębie OUN, wymagających leczenia (głównie choroby rozrostowe) oraz wykazania ewentualnych wrodzonych nieprawidłowości okolicy przysadki (hipoplazja przysadki z ektopią płata tylnego, SOD). Dodatkowo ocenia się wiek biologiczny dziecka na podstawie oceny dojrzałości szkieletu – badanie wieku kostnego, ocenianego metodą Greulicha-Pyle’a. W SNP wiek kostny dziecka jest opóźniony w stosunku do wieku metrykalnego.1,7
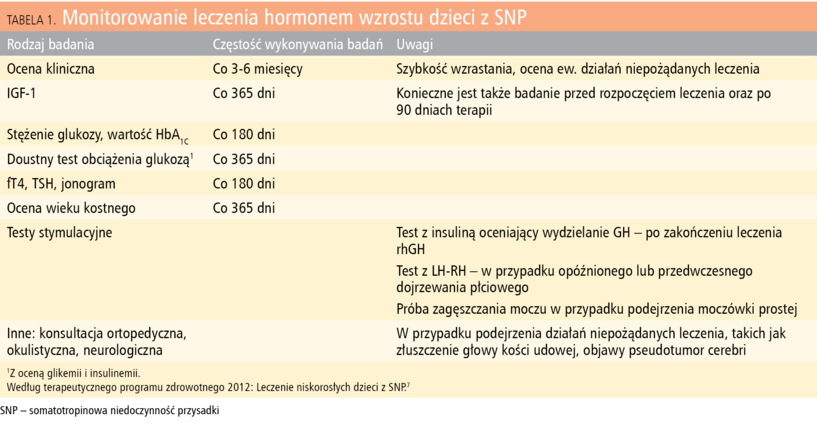
Tabela 1. Monitorowanie leczenia hormonem wzrostu dzieci z SNP
Preparat rhGH podawany jest codziennie wieczorem we wstrzyknięciach podskórnych w dawce: 0,1-0,33 mg/kg/tydzień (0,3-1,0 j./kg/tydzień). Leczenie jest monitorowane podczas wizyt ambulatoryjnych lub krótkotrwałych hospitalizacji (takie leczenie może być w Polsce refundowane do 18 r.ż., nie jest refundowane u dorosłych). Ocenia się skuteczność leczenia (ocena tempa wzrastania) oraz ewentualne działania niepożądane leczenia (m.in. pojawienie się lub progresja istniejących wad postawy). W ramach monitorowania bezpieczeństwa prowadzonej terapii wykonuje się badania kontrolne wymienione w tabeli 1. Do kryteriów wyłączenia z programu należą powikłania terapii w postaci: złuszczenia głowy kości udowej, objawów pseudotumor cerebri(spowodowanych zwiększoną retencją wody), zaburzeń gospodarki węglowodanowej (cukrzyca). Leczenie należy zakończyć w przypadku ujawnienia lub wznowy choroby rozrostowej. Zakończenie terapii promującej wzrastanie następuje wtedy, kiedy wynik leczenia jest niezadowalający oraz w momencie osiągnięcia wieku kostnego powyżej 16 lat przez dziewczynkę i powyżej 18 lat przez chłopca.7 Nie oznacza to jednak konieczności zaprzestania terapii u wszystkich pacjentów. Powinna być ona kontynuowana po zakończeniu procesu wzrastania i wykonaniu testu wydzielania GH po obciążeniu insuliną dla identyfikacji osób wymagających dalszego leczenia w dawkach metabolicznych.
Leczenie niskorosłych dzieci z zespołem Turnera
Zespół Turnera (ZT) uwarunkowany utratą całości lub części materiału genetycznego jednego z dwóch chromosomów X jest jedną z głównych przyczyn niedoboru wzrostu u dziewcząt i należy go brać pod uwagę w diagnostyce różnicowej niedoboru wzrostu u każdej dziewczynki. Zespół charakteryzuje się nieprawidłowym fenotypem, dysgenezją gonad oraz występowaniem wad narządów wewnętrznych, najczęściej układu moczowego i sercowo-naczyniowego, oraz skłonnością do częstszego występowania schorzeń autoimmunologicznych. Zespół występuje z częstością około 1/2500 żywo urodzonych dziewczynek. Dziewczęta z ZT wymagają wielospecjalistycznej opieki medycznej. Obecnie standardem leczenia dziewcząt z ZT, u których występuje niedobór wzrostu, jest stosowanie preparatu rhGH. Ostatnio opublikowane rekomendacje dotyczące postępowania klinicznego u dzieci z zespołem Turnera zostały opracowane przez zespół ekspertów w 2007 r. (Care of Girls and Women with Turner Syndrome: A Guideline of the Turner Syndrome Study Group).11,12
Zakres nieprawidłowości kariotypu w ZT obejmuje m.in.: monosomię chromosomu X (45,X), nieprawidłowości strukturalne chromosomu X, takie jak pierścieniowy chromosom X, izochromosom X oraz różnego typu kariotypy mozaikowe. Rozpoznanie ZT nie obejmuje chłopców z genotypem mogącym odpowiadać ZT, u których rozpoznaje się dysgenezję gonad. U takich chłopców może występować znaczący niedobór wzrostu i mimo dobrej odpowiedzi na leczenie GH obecnie wskazanie to nie jest zawarte w terapeutycznym programie zdrowotnym (lekowym) 2012: Leczenie niskorosłych dzieci z ZT.
W ZT występuje haploidalność genu SHOX (short stature homeobox gene), zlokalizowanego w pseudoautosomalnym regionie chromosomu X, co jest główną przyczyną niedoboru wzrostu u dziewcząt z ZT. Nie stwierdza się u nich bezwzględnego niedoboru GH. Średni wzrost ostateczny jest niższy o ok. 20 cm od średniego wzrostu zdrowej kobiety w danej populacji i wynosi w Polsce ok. 143 cm. Dodatkowe nieprawidłowości układu szkieletowego obejmują m.in.: skrócenie odcinka szyjnego kręgosłupa, koślawość stawów łokciowych i kolanowych, skrócenie IV kości śródręcza. U dziewcząt z ZT występuje zwiększone ryzyko wad postawy, takich jak skolioza czy nadmierna kifoza kręgosłupa (10-20% chorych). W przeciwieństwie do rozwoju somatycznego rozwój intelektualny tych dziewcząt jest na ogół prawidłowy. Z uwagi na częstość występowania i heterogenny obraz kliniczny należy wykluczyć ZT u każdej niskorosłej dziewczynki.11-13
Leczenie rhGH dziewcząt z ZT poprawia tempo wzrastania w dzieciństwie oraz wzrost ostateczny. Terapia ta, stosowana od wczesnych lat 90. XX wieku, w Polsce jest refundowana z budżetu państwa od 1999 roku, obecnie według kryteriów terapeutycznego programu zdrowotnego (lekowego) 2012: Leczenie niskorosłych dzieci z ZT.14 Czynniki predysponujące do osiągnięcia wyższego wzrostu ostatecznego dziewcząt z ZT to: wyższy wzrost na początku leczenia, wysoki wzrost rodziców, wczesne rozpoczęcie leczenia i czas jego trwania oraz dawka rhGH. Nie określono optymalnego momentu rozpoczęcia leczenia rhGH, uznaje się, że celowe jest rozpoczęcie leczenia w momencie zwolnienia tempa wzrastania.12,14 W Polsce leczenie rhGH rozpoczyna się wtedy, gdy u dziewczynki występuje niedobór wzrostu (poniżej 3 centyla).14 Wstępne dane z badania obejmującego najmłodsze dziewczęta z ZT, w wieku od 9 miesięcy do 4 lat, wskazują na skuteczność i bezpieczeństwo leczenia rozpoczętego już w 1 roku życia.15 W pierwszym randomizowanym badaniu przeprowadzonym w Kanadzie i oceniającym wzrastanie dziewcząt z ZT leczonych rhGH porównano ich wzrost ostateczny ze wzrostem historycznej grupy kontrolnej. Wykazano poprawę wysokości ciała dziewcząt (w wieku 7-13 lat) leczonych rhGH w dawce 0,3 mg/kg/tydzień (0,9 j./kg/tydzień) o 7,2 cm w stosunku do grupy kontrolnej w ciągu średnio 5,7 roku.16

Tabela 2. Monitorowanie leczenia hormonem wzrostu dziewcząt z zespołem Turnera