Zalecane dawki rhGH w leczeniu dziewcząt z ZT są większe niż stosowane u dzieci z SNP. W Polsce wynoszą one 0,33-0,47 mg/kg/tydzień (1,0-1,4 j./kg/tydzień).14 Większe dawki niż wymienione nie powodują znaczącej poprawy ostatecznego wzrostu, nie zwiększa się również ryzyko wczesnych działań niepożądanych. Często obserwuje się jednak zwiększone stężenie IGF-1, co stanowi potencjalne ryzyko odległych działań niepożądanych.12,17 Monitorowanie leczenia rhGH polega na ocenie klinicznej i laboratoryjnej (tab. 2). Z uwagi na częstsze występowanie cukrzycy typu 1 i 2 w ZT konieczne jest monitorowanie glikemii oraz utrzymanie prawidłowej masy ciała. Wskazania do zakończenia leczenia to m. in. znaczące zwolnienie tempa wzrastania (<3 cm/rok), osiągnięcie niewyróżniająco wzrostu (≥158 cm) i zaawansowany wiek kostny (>14 lat).14
Leczenie dzieci z zespołem Pradera-Williego
Zespół Pradera-Williego (PWS) występuje z częstością 1/30 000 żywych urodzeń, częstość populacyjna oceniana jest na 1/50 000 i jest najczęstszą genetycznie uwarunkowaną przyczyną otyłości. W przebiegu choroby, poza charakterystycznymi cechami dysmorficznymi, w okresie niemowlęcym występują: hipotonia, trudności w karmieniu, hipogonadyzm (wnętrostwo). W okresie późniejszym występują: narastająca otyłość, niedobór wzrostu, skolioza, hipogonadyzm oraz opóźnienie psychoruchowe i problemy psychologiczne. Za przyczynę nieprawidłowości występujących w PWS uznaje się zaburzenia czynności podwzgórza, o czym świadczą objawy zespołu m.in. obniżone napięcie mięśniowe, upośledzone uczucie sytości, zaburzenia oddychania, zaburzenia termoregulacji, obniżony próg odczuwania bólu, zaburzenia wydzielania neurohormonów, powodujące m.in. upośledzenie procesów wzrastania i hipogonadyzm.18,19
Zespół Pradera-Williego jest chorobą uwarunkowaną genetycznie, w której brakuje ekspresji genów pochodzących od ojca (chromosom 15q11-q13, gen SNRPN). Geny w tym regionie pochodzące od matki są fizjologiczne piętnowane (unieczynnienie genów w procesie metylacji DNA). W 75% przypadków występuje delecja fragmentu ojcowskiego (typ I i II), w 24% disomia uniparenteralna matczyna (UPD), w 1% defekty piętnowania (mikrodelecje w centrum piętnowania, sporadyczne lub dziedziczone), w mniej niż 1% translokacje chromosomu ojcowskiego.18
Dzieci z PWS charakteryzuje łagodne wewnątrzmaciczne opóźnienie wzrastania, średnia urodzeniowa masa ciała wynosi -1,4 SD (-2,8-0,2 SD) a długość ciała -0,5 SD (-2,1-1,4 SD). Niedobór wzrostu nasila się w dzieciństwie i wynosi u dzieci przed rozpoczęciem leczenia rhGH (średni wiek 6,4 roku): -2,2 SD (-4,1-0,3 SD). Wzrost dorosłych wynosi dla kobiet 149-152 cm, a dla mężczyzn 159-162 cm.18 W PWS wykazano zmniejszenie wydzielania GH w testach stymulacyjnych u około 70% chorych, zaburzenia w dobowym wydzielaniu GH i małe stężenia IGF-1. Zaburzenia wydzielania GH i IGF-1, hormonów regulujących m.in. lipolizę i syntezę białek, powodują zaburzenia składu masy ciała chorych na PWS. Leczenie rhGH dzieci z PWS ma zatem na celu nie tylko promocję wzrastania, ale głównie pozytywny efekt metaboliczny GH. U leczonych dzieci obserwuje się poprawę składu masy ciała oraz zwiększenie napięcia i siły mięśniowej. Dzięki temu łatwiejsze jest utrzymanie prawidłowego stanu odżywienia i uzyskanie znacznie lepszych wyników rehabilitacji. U wszystkich dzieci z PWS leczonych rhGH należy zwrócić uwagę na możliwość wystąpienia i/lub nasilenia się skoliozy. Leczenie chorych na PWS musi być kompleksowe, skojarzone z postępowaniem dietetycznym i rehabilitacją.18-24
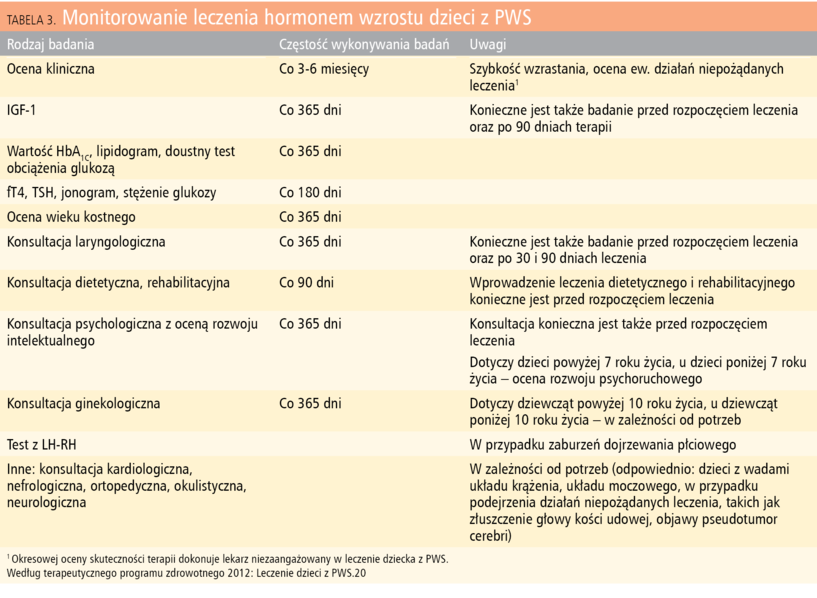
Tabela 3. Monitorowanie leczenia hormonem wzrostu dzieci z PWS
Od 2006 roku w Polsce dzieci z PWS objęte są leczeniem rhGH w ramach terapeutycznego programu zdrowotnego finansowanego przez NFZ (takie leczenie może być refundowane do 18 r.ż., u dorosłych nie jest refundowane).20 Leczenie rozpoczyna się wcześnie, od 2 roku życia, chociaż niektórzy autorzy podają korzystny i bezpieczny wynik terapii rozpoczętej już w 6-12 m.ż. Dawki rhGH wynoszą 0,18-0,47 mg/kg/tydzień (0,54-1,4 j./kg/tydzień). Zalecane jest rozpoczęcie leczenia od dawki mniejszej, a następnie modyfikacja w zależności od wyniku terapii i stężenia IGF-1. U części chorych na PWS obserwuje się zwiększone stężenia IGF-1 w trakcie leczenia, mimo stosowania dość małych w stosunku do rekomendowanych dawek hormonu wzrostu, co może zwiększać ryzyko działań niepożądanych w pierwszych miesiącach leczenia (przerost tkanki limfatycznej nosogardła i ryzyko nasilenia bezdechów nocnych). Wyniki badań wskazują jednak na korzystny długotrwały rezultat leczenia rhGH w zakresie zaburzeń oddychania w czasie snu u chorych na PWS.18,20,25 Kryteria kwalifikacji dziecka z PWS do leczenia rhGH obejmują wiek kostny poniżej 16 lat u dziewcząt i 18 lat u chłopców, stan odżywienia, mierzony wartością wskaźnika masy ciała (BMI), w granicach normy – poniżej 97 centyla dla płci i wieku (wymagana jest co najmniej 6-miesięczna obserwacja w ośrodku prowadzącym terapię rhGH) oraz prawidłowe wyniki badań gospodarki węglowodanowej. Jest to jedyny program leczenia rhGH, w którym niedobór wzrostu nie jest jednym z warunków rozpoczęcia terapii, ponieważ głównym celem leczenia dzieci z PWS jest poprawa zaburzeń metabolicznych występujących w tym schorzeniu. Konieczne jest przeprowadzenie odpowiednich konsultacji, m.in. laryngologicznej, z uwagi na możliwość nasilenia lub wystąpienia zaburzeń oddychania w trakcie snu, oraz wprowadzenie leczenia dietetycznego i rehabilitacyjnego (tab. 3). Do kryteriów wyłączenia z programu, oprócz powikłań będących skutkiem leczenia (zwłaszcza pojawienia się lub nasilenia bezdechów nocnych oraz wystąpienia cukrzycy), należą również: zaniechanie systematycznego leczenia rehabilitacyjnego lub dietetycznego oraz narastanie otyłości, pomimo stosowania leczenia rhGH, dietetycznego i rehabilitacyjnego (wzrost BMI w odniesieniu do norm populacyjnych przyjętych dla wieku i płci o 2 SD lub więcej).20 Istotna wydaje się możliwość kontynuowania leczenia po zakończeniu procesu wzrastania w dawkach działających metabolicznie dla utrzymania osiągniętych wyników leczenia.
Leczenie niskorosłych dzieci z przewlekłą niewydolnością nerek
Przewlekła niewydolność nerek, którą rozpoznajemy przy klirensie kreatyniny poniżej 90 ml/1,73 m2/min, wiąże się z upośledzeniem wzrastania mimo właściwego leczenia choroby zasadniczej i przeszczepienia nerek. Około 50% dzieci, u których PNN rozpoczęła się w dzieciństwie, a leczenie nerkozastępcze rozpoczęto przed 15 rokiem życia, osiąga wyróżniająco niski wzrost ostateczny, poniżej 3 centyla. Wśród przyczyn opóźnienia wzrastania w PNN wymienia się pierwotną chorobę nerek oraz jej powikłania, takie jak niedożywienie, osteodystrofia, niedokrwistość, zaburzenia wodno-elektrolitowe i kwasica. Wydzielanie GH u dzieci z PNN jest prawidłowe i nie zmienia się wraz z obniżaniem przesączania kłębuszkowego, ale występuje nieprawidłowa odpowiedź tkanek na działanie GH. Spowodowane jest to zmniejszoną ekspresją receptora dla GH i zmniejszoną biodostępnością IGF-1. Poprawę wzrastania po zastosowaniu hormonu wzrostu u dzieci z PNN uzyskuje się głównie dzięki normalizacji stosunku IGF-1 do IGFBP-3. W przypadku niskorosłości w przebiegu PNN zastosowanie rhGH jest leczeniem z wyboru i prowadzone jest zarówno w okresie przeddializacyjnym, jak i w trakcie leczenia nerkozastępczego oraz niekiedy po przeszczepieniu nerki.26,27
W Polsce obowiązuje terapeutyczny program zdrowotny (lekowy) 2012: Leczenie niskorosłych dzieci z PNN. Leczenie to prowadzone jest w ośrodkach nefrologicznych. Kryteria kwalifikacji do programu to: niedobór wzrostu (wysokość ciała poniżej -1,88 SD lub tempo wzrastania poniżej -2,0 SD) oraz opóźniony wiek szkieletowy (poniżej 12,5 roku dla chłopców i poniżej 11,5 roku dla dziewczynek) u dziecka z PNN (obniżenie klirensu kreatyniny poniżej 75 ml/1,73 m2/min). Lek podaje się codziennie wieczorem w dawce: 0,33-0,37 mg/kg/tydz. (1,0-1,1 j./kg/tydz.). Kryteria wyłączenia z programu są zbliżone do kryteriów pozostałych programów zdrowotnych (lekowych) leczenia rhGH.27
Podsumowanie
Możliwość stosowania rhGH ma duże znaczenie w leczeniu niskorosłych dzieci kwalifikowanych do terapii według terapeutycznych programów zdrowotnych NFZ, zgodnie z danym rozpoznaniem choroby podstawowej. Leczenie preparatami rhGH jest bezpieczne, jednak wymaga przestrzegania obowiązujących zasad monitorowania terapii. Jest to leczenie nie tylko promujące wzrastanie dziecka, ale także wyrównujące wiele zaburzeń metabolicznych występujących w objętych leczeniem schorzeniach. Należy podkreślić, że dzięki wielokierunkowemu działaniu rhGH możliwa jest znacząca poprawa jakości życia chorych dzieci, w tym korzystny wpływ na ich funkcjonowanie psychospołeczne.