Minisympozjum wrodzone wady metabolizmu w dobie badań przesiewowych
3-metylokrotonyloglicynuria – najczęściej identyfikowana choroba w badaniu przesiewowym noworodków metodą tandemowej spektrometrii mas
Jolanta Sykut-Cegielska,1 Mariusz Ołtarzewski,1 Joanna Taybert,2 Agnieszka Kowalik,1 Dorota Korycińska-Chaaban,2 Ewa Jabłońska1
Słowa kluczowe
3-metylokrotonyloglicynuria, spektrometria mas, deficyt karboksylazy 3-metylokrotonylo-koenzymu A
Wprowadzenie
Dzięki badaniom przesiewowym noworodków metodą tandemowej spektrometrii mas, ostatnio powszechnie wykorzystywanym w diagnostyce rzadkich wrodzonych wad metabolizmu, identyfikowane są liczne przypadki chorób o nieudowodnionym dotychczas znaczeniu klinicznym (w tym 3-metylokrotonyloglicynuria). Często u zidentyfikowanych noworodków nie występują żadne niepokojące objawy w dłuższym okresie obserwacji. W związku z tym pojawiają się wątpliwości co do konieczności wprowadzenia interwencji medycznej w postaci: zmiany diety dziecka (ograniczenie białka naturalnego, szczególnie leucyny), farmakoterapii (suplementacja L-karnityny), stałych zaleceń unikania przedłużonego głodzenia i natychmiastowego działania w sytuacjach wzmożonego katabolizmu (zagrożenie dekompensacją metaboliczną). Niezależnie od powyższych zaleceń większość klinicystów potwierdza potrzebę monitorowania przebiegu choroby mimo pojawiających się zarzutów dotyczących medykalizacji.1
Co to jest 3-metylokrotonyloglicynuria (deficyt MCC)?
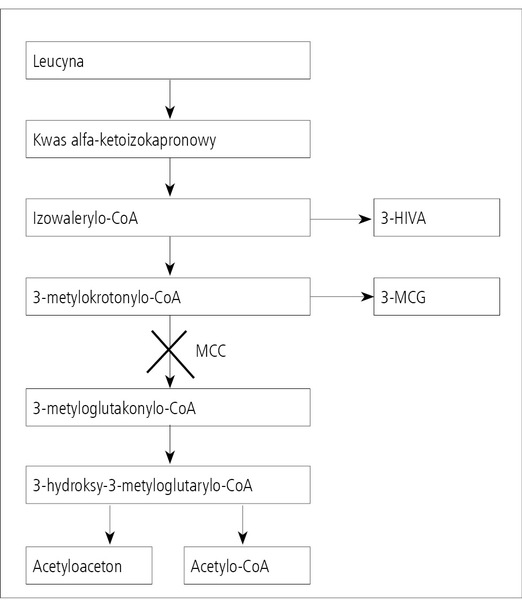
Rycina 1. Schemat przemiany leucyny z zaznaczeniem miejsca deficytu MCC.
3-metylokrotonyloglicynuria (3-MCG) jest względnie nową jednostką chorobową, po raz pierwszy opisaną w 1984 roku,2 choć wcześniej publikowano opisy pojedynczych przypadków jeszcze bez identyfikacji defektu metabolicznego. Nazwą 3-metylokrotonyloglicynuria określa się zjawisko zwiększonego wydalania w moczu 3-metylokrotonyloglicyny, któremu towarzyszy wydalanie kwasu 3-hydroksyizowalerianowego (3-HIVA) i ew. tiglylglicyna, badane metodą chromatografii gazowej sprzężonej ze spektrometrią mas (GC/MS). W rzadkich przypadkach w badaniu metodą GC/MS jedynym nieprawidłowym metabolitem wykrywanym w moczu jest 3-HIVA bez (uważanego do niedawna za patognomoniczne) wzmożonego wydalania MCG.3 Markerem biochemicznym z krwi jest zwiększone stężenie 3-hydroksyizowalerylokarnityny (C5OH) wykrywane w suchej kropli krwi. Jest to spowodowane izolowanym deficytem karboksylazy 3-metylokrotonylo-koenzymu A (3-methylcrotonyl-CoA carboxylase deficiency, MCC deficiency). Enzym MCC bierze udział w przemianie jednego z aminokwasów rozgałęzionych, tj. leucyny; umożliwia przekształcenie 3-metylokrotonylo-koenzymu A w 3-metyloglutakonylo-koenzym A (ryc. 1). Jest jedną z czterech karboksylaz uczestniczących w cyklu metabolizmu biotyny, potencjalnie jest zatem enzymem biotynozależnym. Potwierdzenie deficytu enzymu MCC u pacjentów bez zwiększonego wydalania MCG było podstawą do wysunięcia sugestii o zmianie nazwy z 3-MCG na deficyt MCC.3 Deficyt MCC dziedziczy się w sposób autosomalny recesywny. Odpowiedzialne są mutacje w genie MCCC1 (dawniej MCCA) kodującym podjednostkę alfa enzymu lub MCCC2 (dawniej MCCB) kodującym podjednostkę beta tego enzymu. Gen MCCC1 składa się z 19 eksonów i jest zlokalizowany na chromosomie 3q25-q27, zaś MCCC2 o 17 eksonach – 5q12-q13.4 Wśród dotychczas wykrytych w tych genach są mutacje o typie nonsense, missense, frameshift, delecji, małych insercji oraz splice site.5
Rozpoznawanie i fenotyp deficytu MCC przed erą badań przesiewowych noworodków
Jeszcze do niedawna deficyt MCC uznawany był za jedną z klasycznych acydurii organicznych z grupy rzadkich wrodzonych wad metabolizmu pośredniego aminokwasów rozgałęzionych, które w wyniku bloku enzymatycznego przebiegają z gromadzeniem kwasów karboksylowych, co powoduje objawy przypominające ostre zatrucie, czyli tzw. zespół intoksykacji. Do obrazu klinicznego deficytu MCC należały głównie nawracające epizody dekompensacji klinicznej (związanej ze stanami wzmożonego katabolizmu lub nadmiernego obciążenia białkiem) oraz dysfunkcja wątroby w postaci zespołu Reye’opodobnego, hipoglikemia i objawy neurologiczne (hipotonia, drgawki, dystonia, epizody udaropodobne, leukodystrofia, opóźnienie rozwoju psychoruchowego).6-8 Charakterystyczny dla choroby jest też szczególny zapach wyczuwalny od osób chorych, przypominający zapach moczu kocura. Po ustaleniu rozpoznania postępowanie polegało na stosowaniu diety z ograniczeniem białka naturalnego wykorzystującej środki spożywcze specjalnego przeznaczenia medycznego niezawierające leucyny, z zaleceniem zapewnienia podaży kalorycznej w stanach wzmożonego katabolizmu oraz suplementacji karnityny w przypadku stwierdzenia jej niedoboru.
Rozpoznawanie i fenotyp deficytu MCC obecnie
W ostatnim czasie badania przesiewowe noworodków metodą tandemowej spektrometrii mas (tandem MS, MS/MS) ujawniły inne od dotychczas znanego oblicze tej wrodzonej wady metabolizmu. Okazało się, że zwiększone (wykraczające poza zakres wartości referencyjnych) stężenie C5OH wykrywane w suchej kropli krwi stanowi bardzo często stwierdzane odchylenie w badaniu przesiewowym; według Baumgartnera i wsp. u 1 na 50 000 noworodków,9 według Arnold i wsp. u 1 na 36 000 noworodków.1 Jednocześnie zakres obserwowanych objawów deficytu MCC znacznie się poszerzył – od ciężkich postaci noworodkowych do braku objawów nawet do wieku dorosłego. Różnorodność obrazu klinicznego (fenotypu) może wystąpić nawet w obrębie tej samej rodziny, mającej zatem ten sam genotyp. W pracy Grünert i wsp. wśród 8 pacjentów zdiagnozowanych jako cierpiących na deficyt MCC w ramach diagnostyki rodzinnej u 3 nie występowały objawy, u kolejnych 3 objawy były obecne, na temat pozostałych 2 brakuje danych.10 Wśród objawów klinicznych stwierdzano: opóźnienie rozwoju psychoruchowego, opóźnienie rozwoju mowy, obniżenie napięcia mięśniowego, nadpobudliwość ruchową i niechęć do jedzenia mięsa. W ramach diagnostyki rodzinnej czteroletniego chłopca z deficytem MCC (hipotonią i miernym opóźnieniem rozwoju ruchowego) wykryto to zaburzenie u jego 17-letniego brata bez objawów, a w przypadku chłopca z drgawkami atonicznymi i kwasicą metaboliczną stwierdzonymi w wieku niemowlęcym (zdiagnozowany w ramach skriningu selektywnego) – u 3-letniego brata z opóźnieniem rozwoju mowy i makrocefalią potwierdzono deficyt MCC.
Visser i wsp. opisali występowanie deficytu MCC u 7-tygodniowej dziewczynki z kardiomiopatią rozstrzeniową, jej 10-letniego brata z niepełnosprawnością intelektualną, ale też u ich zdrowego ojca. Rozpoznanie u dwóch ostatnich ustalono w ramach diagnostyki rodzinnej po wykryciu nieprawidłowości u niemowlęcia. W konkluzji jednak autorzy sugerowali, że w tej rodzinie nie można wykluczyć odmiennego genotypu u poszczególnych członków rodziny.11
Ustalenie ostatecznego rozpoznania dodatkowo utrudnia możliwość wystąpienia odchylenia pochodzenia odmatczynego wykrywanego u noworodka metodą MS/MS. Wiadomo bowiem, że matka z deficytrm MCC (zwykle o nim niewiedząca) karmiąca piersią przekazuje dziecku metabolity, które są identyfikowane w badaniu przesiewowym. Z czasem, gdy dieta niemowlęcia jest rozszerzana o inne pokarmy, wynik badania profilu acylokarnityn się normalizuje. W ten sposób badania przesiewowe noworodków metodą MS/MS pozwalają na wykrycie wrodzonej wady metabolizmu nie tylko u noworodków, ale czasem u ich dorosłych matek (materiał własny).10,12 Są one mimo częstego braku danych w wywiadzie o niepokojących objawach (przebieg bez- lub skąpoobjawowy) zagrożone ujawnieniem klinicznym deficytu MCC w sytuacjach związanych ze wzmożonym katabolizmem (operacja, ciężka infekcja, przedłużone głodzenie). U niektórych z nich stwierdzano wcześniej stany dekompensacji metabolicznej związane z infekcjami (materiał własny).10
Przeanalizowano przebieg choroby w grupie 14 pacjentów zidentyfikowanych w badaniach przesiewowych noworodków (w wieku 1,75-6,5 roku), jak również ich czworga rodzeństwa (w wieku 4,5-12,25 roku) oraz 7 matek (w wieku 22-38 lat) zdiagnozowanych w trakcie poszerzonych badań rodzinnych.13 Wszyscy byli bez objawów poza jednym dzieckiem z opóźnionym rozwojem mowy i 2 z wywiadem nietolerancji wysiłku fizycznego i osłabieniem kończyn. Autorzy dokonali analizy 37 przypadków opublikowanych wcześniej, z której wynikało, że 27% przypadków było bezobjawowych, 11% dzieci zmarło, 32% miało opóźniony rozwój psychoruchowy, 16% drgawki i 49% niespecyficzne objawy neurologiczne. Na podstawie powyższych danych wysunięto sugestię, że być może nie wszystkie objawy mogły być związane z deficytem MCC, a zwiększone stężenie C5OH w badaniu przesiewowym może nie mieć znaczenia klinicznego. Przeciwną opinię wyrażono w późniejszym opracowaniu Grünert i wsp.10 Dokonano analizy danych klinicznych, biochemicznych, enzymatycznych i wyników badań molekularnych w grupie 88 pacjentów z rozpoznanym deficytem MCC, w tym 53 (60%) zidentyfikowanych w badaniach przesiewowych noworodków, 26 (30%) zdiagnozowanych wobec wystąpienia objawów klinicznych lub w ramach badań rodzinnych oraz 9 (10%) matek (w wieku 24-38 lat) zidentyfikowanych w trakcie diagnostyki różnicowej nieprawidłowości stwierdzanych u noworodków. Zanotowano trzy zgony. Pierwszy z powodu nagłego zatrzymania akcji serca w 33 dniu życia u dziecka zidentyfikowanego w ramach badań przesiewowych noworodków, z ciężką dekompensacją metaboliczną od pierwszego dnia życia, hipoglikemią, hiperamonemią i kwasicą mleczanową. Drugi zgon nastąpił u 6-tygodniowego niemowlęcia z uogólnioną wiotkością, drgawkami mioklonicznymi i ostrą dekompensacją metaboliczną. Trzeci zgon dotyczył 8-letniej dziewczynki, u której jednak objawy ze strony serca (tachykardia komorowa i nagłe zatrzymanie akcji serca) wydają się raczej spowodowane wykrytymi mutacjami w genie dla receptora rjanodyny RyR2, odpowiadającymi za nieprawidłowy napływ wapnia w komórkach mięśnia sercowego, tj. chorobą niezwiązaną przyczynowo z deficytem MCC. U 34 z 80 pacjentów (tj. w ponad 40%; w 8 przypadkach brakuje danych) występowały objawy kliniczne, od ciężkich dekompensacji metabolicznych (z encefalopatią, ketokwasicą i hipoglikemią) do objawów mięśniowych, upośledzonego rozwoju, deficytu uwagi i nadpobudliwości. W grupie 53 dzieci zdiagnozowanych w ramach badań przesiewowych noworodków 36 nie miało objawów, a u 13 stwierdzano różne objawy kliniczne (w czterech przypadkach brakuje danych). Oznacza to, że prawie co czwarte dziecko zidentyfikowane w fazie przedobjawowej ujawniło objawy kliniczne deficytu MCC. W tym w pięciu przypadkach wystąpiły pełnoobjawowe incydenty dekompensacji metabolicznej. Co ciekawe, w przypadkach wykrytego deficytu MCC pochodzenia odmatczynego u jednej z matek występowało przewlekłe zmęczenie, a poza tym nie stwierdzano żadnych niepokojących objawów, u drugiej jednak występowały nawracające dekompensacje metaboliczne w przebiegu infekcji gorączkowych, a także kardiomiopatia, parestezje i epizod udaru mózgu.
Własne wyniki analizy przypadków rozpoznanych w kraju wykazały dużą wykrywalność deficytu MCC w ramach badań przesiewowych noworodków, tj. metodą MS/MS.14 Deficyt MCC rozpoznano ogółem u 42 osób, w tym na podstawie skriningu selektywnego wykryto 11 przypadków, diagnostyki rodzinnej – 5 przypadków, zaś w badaniach przesiewowych noworodków zidentyfikowano 19 dzieci i 7 matek. Poza grupą pacjentów zdiagnozowanych w ramach skriningu selektywnego i jedną z matek (u której w wywiadzie był przebyty w wieku 9 lat zespół Reye’a o nieustalonej wówczas etiologii) nie obserwowano niepokojących objawów. Najdłuższy okres obserwacji pacjentów zidentyfikowanych w przesiewowych badaniach noworodków wynosił jednak tylko 10 lat, a najpóźniejszy wiek ujawnienia się deficytu MCC w analizowanej grupie pacjentów z objawami to 12 lat.
Wśród 11 pacjentów z objawami zwracały uwagę: wymioty (u 4), drgawki lub ich ekwiwalenty (u 5), opóźnienie rozwoju psychoruchowego (u 3) i inne objawy neurologiczne. Hipoglikemię stwierdzono tylko u jednego pacjenta.
Stężenie C5OH-karnityny w suchej kropli krwi w momencie rozpoznania wynosiło 0,72-41,0 μmol/l u pacjentów z objawami i 0,51-31,5 μmol/l (wartości referencyjne <0,47 μmol/l) u pacjentów poddanych badaniom przesiewowym. Znaczący wzrost C5OH w suchej kropli krwi (>3,00 umol/l) stwierdzono odpowiednio w 45 i 48% przypadków zdiagnozowanych w skriningu selektywnym i przesiewowym badaniu noworodkowym. W chwili rozpoznania, niezależnie od sposobu diagnostyki, wydalanie 3-HIVA i MCG następowało u wszystkich pacjentów z wyjątkiem jednego, u którego pozostawało niewykrywalne przez 11 kolejnych miesięcy dalszej obserwacji. Masywne zwiększenie stężenia 3HIVA i MCG, definiowane jako większe niż 500 mmol/mol kreatyniny i 200 mmol/mol kreatyniny (odpowiednio wartości referencyjne <20 i niewykrywalne), było stwierdzane u około połowy pacjentów diagnozowanych zarówno w diagnostyce objawowej, jak i przedobjawowej.
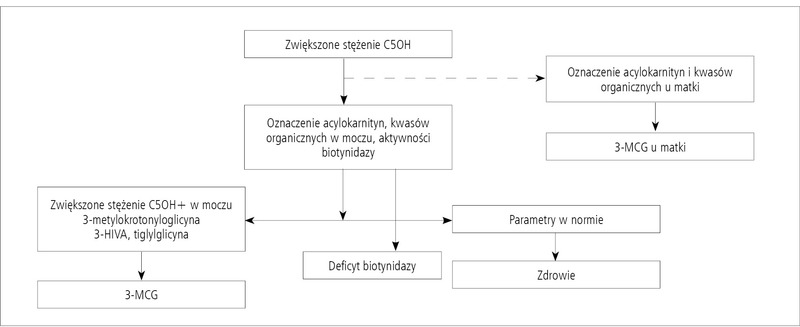
Rycina 2. Schemat postępowania w przypadku zwiększonego stężenia C5OH w MS/MS.
U matek z deficytem MCC zwracało uwagę znaczne zwiększenie stężenia C5OH w suchej kropli krwi w zakresie 10,2-54,1 µmol/l.
Wobec skomplikowanego procesu diagnostycznego w przypadku deficytu MCC autorzy proponują korzystanie z algorytmu diagnostycznego (ryc. 2), który powinien być pomocny w procesie różnicowania ewentualnych przyczyn zwiększonego stężenia C5OH u noworodka.

Tabela. Pierwszoplanowe badania w przypadku zwiększonego stężenia C5OH w badaniu przesiewowym noworodków