Jako przyczynę bierze się pod uwagę przewagę kwasów omega-6, szczególnie w emulsjach na bazie soi, oraz zawartość fitosteroli, które mogą mieć znaczenie w rozwoju cholestazy u wcześniaków11,12.
Emulsje oparte na oleju z ryb, zawierające głównie kwasy tłuszczowe omega-3, wydają się z kolei mieć znaczenie protekcyjne i wspomagają powrót do zdrowia, ale dowody nie są jednoznaczne5,12.
Częstość występowania atrezji dróg żółciowych u wcześniaków jest podobna jak u noworodków urodzonych w terminie. Może dotyczyć nawet 7,1% dzieci urodzonych <37 t.c.13. Ze względu jednak na znacznie częstsze występowanie cholestazy u wcześniaków w przebiegu stosowanego żywienia pozajelitowego, opóźnionego wprowadzania żywienia enteralnego czy nawracających zakażeń diagnoza atrezji dróg żółciowych w tej grupie pacjentów jest zwykle stawiana z opóźnieniem. Być może mają na to wpływ zalecenia z 2015 roku dotyczące wskazań do biopsji wątroby czy badania scyntygraficznego dróg żółciowych, które należy odroczyć do czasu uzyskania przez dziecko wieku postkoncepcyjnego co najmniej 40 tygodni oraz masy ciała >2 kg i rozważyć przede wszystkim u pacjentów, u których stwierdza się występowanie acholicznych stolców3,5,10.
Ważnym zagadnieniem w etiologii cholestazy u noworodka jest tzw. idiopatyczne zapalenie wątroby. Jest ono przyczyną 25-30% przypadków cholestazy5. Stan ten nie ma określonej etiologii i najczęściej stanowi diagnozę z wykluczenia innych znanych przyczyn, częściej u niemowląt urodzonych przedwcześnie. Cholestaza na ogół rozwija się w kilka tygodni po urodzeniu i może współwystępować z hepatomegalią, łagodnym wzrostem aktywności aminotransferaz, normalnym lub małym stężeniem GGTP. Acholiczne stolce są rzadkie i jeśli występują, mają niekorzystną wartość prognostyczną. Rokowanie jest zazwyczaj pomyślne, z ponad 90% kliniczną i biochemiczną normalizacją do 1 roku życia, przy niewielkim ryzyku rozwinięcia przewlekłej choroby wątroby w przypadkach rodzinnych5.
Obecnie postęp w diagnostyce umożliwiający wykorzystanie m.in. badań genetycznych (np. NGS – next generation sequencing) pozwala na stopniowe zmniejszanie liczby stawianych rozpoznań cholestazy pod postacią tzw. idiopatycznego zapalenia wątroby1.
Diagnostyka
Ze względu na konieczność przeprowadzenia szerokiej diagnostyki różnicowej ważne jest dobre rozplanowanie badań. Priorytetem powinno być rozpoznanie stanów wymagających pilnej interwencji4. Nawet wtedy jednak, gdy specyficzne leczenie jest niedostępne, noworodki, które mają cholestazę, mogą odnieść korzyści z wczesnego rozpoznania i wdrożenia leczenia objawowego w celu zapobiegania powikłaniom i uniknięcia wykonywania niepotrzebnych badań. Podsumowując, ważne są zarówno czas, jak i kolejność wykluczania chorób5.
Niestety nie zawsze zasady te funkcjonują w praktyce. Powodami tej sytuacji są m.in. wczesny wypis dziecka z oddziału noworodkowego bez odpowiedniego monitorowania stężenia bilirubiny w surowicy po wypisie, wprowadzająca w błąd interpretacja koloru stolca czy błędna diagnoza żółtaczki skojarzonej z karmieniem piersią5.
Ważnym etapem diagnostycznym jest dobrze zebrany wywiad lekarski. Należy zwrócić w nim uwagę na pokrewieństwo rodziców, występowanie cholestazy w rodzinie (rodzeństwo, rodzice) oraz historię położniczą matki, w tym poronienia, występowanie cholestazy czy infekcji w trakcie trwania ciąży1.
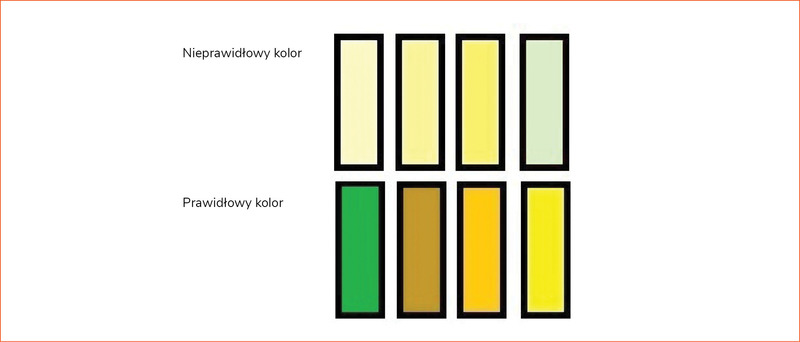
Rycina 1. Przykłady koloru stolca*
Badanie przedmiotowe odgrywa istotną rolę w ocenie dziecka z przedłużającą się żółtaczką. Należy zwrócić szczególną uwagę na hepatosplenomegalię oraz zabarwienie stolca (zalecenia 1A według ESPGHAN)1, które przedstawiono na rycinie 114.
Obraz kliniczny cholestazy u noworodków może się różnić w zależności od etiologii. Należy pamiętać, że w ciągu pierwszych tygodni życia stężenie bilirubiny pośredniej zazwyczaj się zmniejsza, dając fałszywe wrażenie, że żółtaczka ustępuje.
Obecność acholicznych stolców jest zawsze niepokojąca, ale niediagnostyczna dla niedrożności dróg żółciowych. Może również występować w ciężkiej cholestazie wewnątrzwątrobowej5. Z drugiej strony obecność zabarwionych stolców sugeruje drożność zewnątrzwątrobowych dróg żółciowych, co pozwala zmniejszyć podejrzenie atrezji. Jednak we wczesnym przebiegu atrezji stolce mogą pojawić się normalnie lub naprzemiennie zabarwione, ważne zatem, aby ich kolor był oceniany w czasie. Z kolei ciemny mocz jest powszechny i traktowany jako niespecyficzny wskaźnik sprzężonych hiperbilirubinemii, ale nie jest patognomoniczny.
Niektóre niemowlęta mogą mieć koagulopatię wtórną do niedoboru witaminy K wynikającego z jej złego wchłaniania. Może się ona objawiać jako krwawienia z przewodu pokarmowego, z pępka, a nawet jako krwawienie śródczaszkowe. Taka postać wymaga leczenia witaminą K. Koagulopatia może być również spowodowana niewydolnością wątroby, co może wskazywać na ciężkie uszkodzenie tego narządu w przebiegu cholestazy.
Splenomegalia współwystępuje z hepatomegalią w blisko połowie przypadków5. Szczególnie dotyczy to niemowląt z infekcją wrodzoną, marskością wątroby i nadciśnieniem wrotnym, w przebiegu chorób spichrzeniowych czy zaburzeń hemolitycznych. W początkowym okresie pozawątrobowej niedrożności dróg żółciowych śledziona ma zazwyczaj normalny rozmiar.
Wyczuwalna masa w prawym górnym kwadrancie brzucha może wskazywać na torbiel dróg żółciowych1,5.
W przypadku infekcji z grupy TORCH należy zwrócić uwagę na obecność małogłowia i hipotrofii wewnątrzmacicznej1,5.
Dysmorficzne rysy twarzy mogą sugerować zaburzenia chromosomalne czy też zespół Alagille’a. W tym ostatnim przypadku należy szczególnie uwzględnić występowanie szmeru nad sercem i rozważyć wykonanie badania echo serca (najczęściej występuje tu obwodowe zwężenie tętnic płucnych). W przypadku podejrzenia zakażeń wrodzonych czy zespołu Alagille’a ważna jest także ocena okulistyczna.
W diagnostyce biochemicznej pierwszym krokiem w ustaleniu rozpoznania cholestazy u noworodków jest laboratoryjne potwierdzenie sprzężonej hiperbilirubinemii, która jest jej bezpośrednim markerem. Towarzyszącym standardem jest ocena parametrów funkcji wątroby poprzez określenie aktywności enzymów: aminotransferazy alaninowej (AlAT – alanine aminotransferase), aminotransferazy asparaginianowej (AspAT – aspartate aminotransferase), fosfatazy alkalicznej (AP – alkaline phosphatase), γ-glutamylotranspeptydazy (GGTP) oraz układu krzepnięcia: czasu protrombinowego (PT – prothrombin time) i międzynarodowego współczynnika znormalizowanego (INR – international normalized ratio) oraz poziomu albumin. Wartość GGTP jest zazwyczaj wyższa u noworodków niż u starszych dzieci. Należy jednak pamiętać, że niektóre choroby mogą występować z prawidłowym lub małym stężeniem GGTP, takie jak: postępująca rodzinna cholestaza wewnątrzwątrobowa (PFIC – progressive familial intrahepatic cholestasis) typu 1 i 2, zaburzenia syntezy kwasów żółciowych, zespół ARC (arthrogryposis – artrogrypoza, renal tubular dysfunction – zaburzenia czynności cewek nerkowych, cholestasis – cholestaza)9. Oznaczenie stężenia AP jest mniej przydatne u noworodków, biorąc pod uwagę duże różnice w prawidłowym poziomie tego enzymu u młodych niemowląt1. AP jest ponadto wydzielana także przez kości, jelito cienkie i nerki9. Z kolei izolowane podwyższone stężenie AspAT bez wzrostu AlAT i bilirubiny może wskazywać na proces pozawątrobowy (mięśniowy lub hematologiczny)1,8.