IV edycja Kongresu HPV już 12-13 czerwca! Poznaj najnowsze trendy w profilaktyce, diagnostyce i leczeniu raka szyjki macicy i innych schorzeń związanych z HPV | Sprawdź >
Abc chorób metabolicznych
Odyseja diagnostyczna pacjentów z chorobą Sanfilippa
lek. Monika Limanówka1
prof. dr hab. n. med. Jolanta Wierzba2

lek. Monika Limanówka
- Mukopolisacharydoza typu III (MPS III) – postępująca choroba neurodegeneracyjna
- Przebieg i obraz kliniczny choroby Sanfilippa
- Diagnostyka, postępowanie i rokowanie u pacjentów z MPS III
Lizosomalne choroby spichrzeniowe to grupa ponad 50 defektów metabolicznych uwarunkowanych genetycznie. Wszystkie są chorobami monogenowymi dziedziczonymi, z nielicznymi wyjątkami, w sposób autosomalny recesywny. U podłoża każdej z nich leży niedobór enzymu odpowiedzialnego za degradację produktów metabolizmu komórkowego, który skutkuje akumulacją nierozłożonego produktu w lizosomach i tym samym upośledza ich funkcję.
Przez wiele lat lizosomy uważane były za niezbyt istotną organellę komórkową mającą za zadanie utylizację produktów przemian wewnątrzkomórkowych i odzyskiwanie cząsteczek budulcowych potrzebnych do dalszych przemian. Na przestrzeni ostatnich lat pogląd na rolę lizosomu w komórce bardzo się zmienił. W wielu badaniach dowiedziono, że ta organella stanowi centrum koordynujące wiele ścieżek sygnałowych w komórce, w tym jest kluczowym sensorem wewnątrzkomórkowych stężeń substancji odżywczych związanych ze szlakiem mTOR. W komórce odbywa się nieustanny obrót białek, a lizosomy zapewniają koordynację procesów degradacji i autofagii białek w zależności od potrzeb poszczególnych organelli1.
Defekt metaboliczny dotyczący enzymów lizosomalnych skutkuje zaburzeniami procesów na różnych poziomach. Spichrzanie nierozłożonego produktu jest najbardziej oczywistym, pierwszym etapem choroby. To, co dzieje się później, to kaskada zdarzeń prowadząca do upośledzenia funkcji całej komórki oraz jej otoczenia, gdyż wadliwie działająca komórka zaczyna produkować cytokiny prozapalne. Następnym etapem jest apoptoza komórki, jej rozkład w tkankach przez komórki fagocytarne, w których w dalszym ciągu dochodzi do akumulacji substratu i w związku z tym wydzielania cytokin prozapalnych w jeszcze większej ilości. Do tego następuje także wtórne zahamowanie rozkładu innych substratów w lizosomach, co dodatkowo wzmacnia proces zapalny.
Mukopolisacharydoza (MPS) typu III jest najczęściej rozpoznawanym typem mukopolisacharydozy, występuje z częstością mniej więcej 1 na 100 000 urodzeń. Jako pierwszy chorobę tę opisał w 1963 roku amerykański lekarz Sylwester Sanfilippo, od którego nazwiska nazywana jest chorobą Sanfilippa.
W przypadku MPS III dochodzi do zaburzenia degradacji siarczanu heparanu (HS – heparan sulfate) wewnątrz lizosomów. HS to polisacharyd o ujemnym ładunku (inaczej glikozoaminoglikan [GAG]), który przyłączany jest do ogromnej liczby białek znajdujących się zarówno na powierzchni, jak i w wewnątrzkomórkowym matriksie. HS w połączeniu z białkami tworzy proteoglikany, które katabolizowane są w lizosomach. W procesie tym bierze udział kilka enzymów, w związku z czym wyróżniamy 5 typów MPS III (A-E) zależnie od rodzaju uszkodzonego enzymu2 (tab. 1).
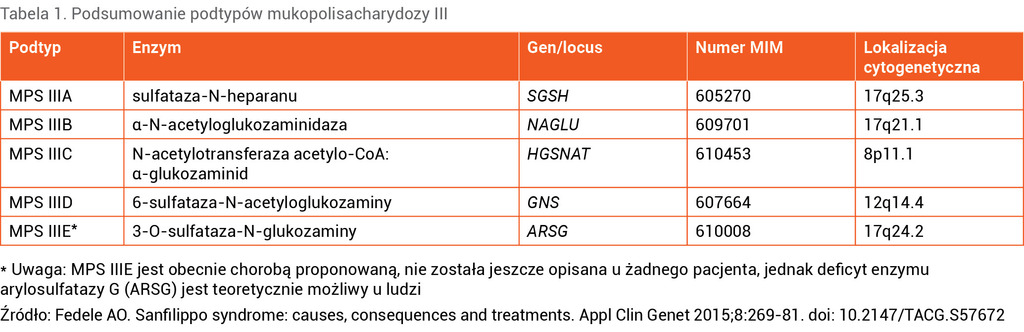
Tabela 1. Podsumowanie podtypów mukopolisacharydozy III
Akumulacja HS w ośrodkowym układzie nerwowym (OUN) powoduje lawinę wtórnych procesów patofizjologicznych, takich jak: reakcja zapalna i apoptoza neuronów, astocytoza, mikroglioza oraz dezorganizacja synaptyczna3.
Dokładny przebieg zdarzeń od momentu kumulacji HS do degeneracji OUN jest stosunkowo słabo poznany. Odkryto, że kumulacja HS hamuje wtórnie degradację gangliozydów GM2 i GM3, tym samym doprowadzając do wtórnego spichrzania tych związków zarówno w sposób bezpośredni, jak i poprzez negatywny wpływ HS na regulację syntezy gangliozydów. Wpływ akumulacji gangliozydów GM2 i GM3 na proces neurodegeneracji w przebiegu MPS III nadal jest przedmiotem badań, najbardziej prawdopodobny wydaje się jednak silny wpływ tych substancji na odpowiedź zapalną, która w znacznym stopniu przyspiesza degenerację neuronów OUN. Dodatkowo dowiedziono na modelach zwierzęcych, że w MPS III dochodzi w większym stopniu do spichrzania zarówno GAG, jak i gangliozydów w komórkach nerwowych niż w innych typach MPS4.
Przebieg kliniczny MPS III
Pogląd na przebieg kliniczny choroby Sanfilippa zmienił się w ostatnich latach. Dzięki powszechności diagnostyki genetycznej, zwłaszcza badań panelowych oraz całoegzomowego sekwencjonowania genomu (WES – whole-exome sequencing), coraz częściej rozpoznanie to jest ustalane u pacjentów z łagodniejszą postacią choroby, u których nie występują charakterystyczne cechy fenotypowe.
Tak jak każda lizosomalna choroba spichrzeniowa MPS III to spektrum fenotypów zależnych od aktywności resztkowej enzymu. Dla ułatwienia fenotypy te nazywane są: postacią ciężką, postacią łagodną lub postacią o późnym początku (attenuated form).
Najcięższa postać choroby jest związana z najmniejszą aktywnością resztkową enzymu (zwykle bliską zera). Jej przebieg można podzielić na 3 fazy poprzedzone okresem bezobjawowym trwającym pierwszych kilka lub kilkanaście miesięcy życia. W pierwszej fazie, która trwa od 1 do 3 roku życia dziecka, obserwuje się powolne zahamowanie rozwoju psychoruchowego (najbardziej wyraźne w sferze mowy, funkcje ruchowe są początkowo w pełni zachowane). Charakterystyczne objawy to: różnie nasilona dysmorfia (pogrubiałe rysy twarzy, synophrys, nadmierne owłosienie), powiększenie narządów wewnętrznych, sztywność stawowa. W pierwszych latach choroby dominują zaburzenia zachowania, które stopniowo ustępują wraz z postępem degeneracji OUN. W drugiej fazie choroby, która zaczyna się w 3-4 roku życia, obserwuje się: postępujące pogorszenie funkcji poznawczych, nasilenie zaburzeń zachowania i zaburzenia snu. Trzecia faza zaczyna się zwykle w wieku nastoletnim i objawia zaawansowanym otępieniem oraz utratą funkcji motorycznych. Problemy z zachowaniem stopniowo ustępują. Chorzy stają się całkowicie zależni od innych osób, tracą możliwość samodzielnego poruszania się, dołączają dysfagia i porażenie spastyczne, stan pacjentów określa się jako wegetatywny. Zgon następuje w drugiej dekadzie lub na początku trzeciej dekady życia5.
U pacjentów z pośrednią postacią choroby na pierwszy plan wysuwa się opóźnienie rozwoju z często towarzyszącymi zaburzeniami zachowania (takimi jak: nadpobudliwość, wybuchy złości, zachowania destrukcyjne, agresja). Opóźnienie rozwoju mowy zauważane jest około 4 roku życia. Zaburzenia zachowania podobnie jak zaburzenia snu występują niestale, zdarzają się okresy prawidłowego funkcjonowania. W tej grupie chorych w wywiadzie dominują nawracające infekcje dróg oddechowych we wczesnych latach życia. Hepatomegalia także nie jest charakterystyczna dla tej postaci choroby
W przypadku dorosłych osób powodem rozpoczęcia diagnostyki genetycznej były takie schorzenia, jak dystrofia siatkówki lub przerostowa kardiomiopatia, pacjenci ci nie prezentowali zaburzeń funkcji poznawczych6. Jest to przykład najłagodniejszego przebiegu klinicznego MPS III. W piśmiennictwie można znaleźć opisy przypadków pacjentów z demencją zdiagnozowanych w szóstej, a nawet w ósmej dekadzie życia, którzy prawidłowo funkcjonowali przez lata7.
Wydaje się, że wielu pacjentów z postacią o późnym początku nigdy nie zostało zdiagnozowanych, natomiast szczegółowe dane byłyby dostępne dopiero po wprowadzeniu przesiewowych badań noworodków w kierunku MPS III.