Nowe leki w terapii cukrzycy typu 2
Ralph A. DeFronzo, MD, Curtis L. Triplitt, PharmD, CDE, Muhammad Abdul-Ghani, MD, Eugenio Cersosimo, MD
W skrócie
Głównymi zaburzeniami odpowiedzialnymi za rozwój i progresję cukrzycy typu 2 są upośledzone wydzielanie insuliny, zwiększone wytwarzanie glukozy w wątrobie oraz zmniejszone zużycie glukozy w tkankach obwodowych. Patofizjologia tej choroby obejmuje jednak również oporność adipocytów na insulinę (zwiększoną lipolizę), zmniejszenie wydzielania inkretyn lub wrażliwości na ich działanie, zwiększenie wydzielania glukagonu, zwiększone zwrotne wchłanianie glukozy w nerkach oraz insulinooporność/dysfunkcję neuroprzekaźników w mózgu. Mimo że obecnie w leczeniu cukrzycy koncentrujemy się na zmniejszaniu stężenia glukozy we krwi, celem leczenia powinno być opóźnienie progresji choroby, a w końcu utraty skuteczności działania leków. Ostatnio wprowadzone innowacyjne metody leczenia są ukierunkowane na liczne defekty patofizjologiczne występujące w cukrzycy typu 2. Optymalne postępowanie powinno polegać na wczesnym rozpoczęciu skojarzonego leczenia wieloma lekami o różnych mechanizmach działania. W niniejszym przeglądzie omówiono nowe możliwości leczenia, które wydają się szczególnie obiecujące.
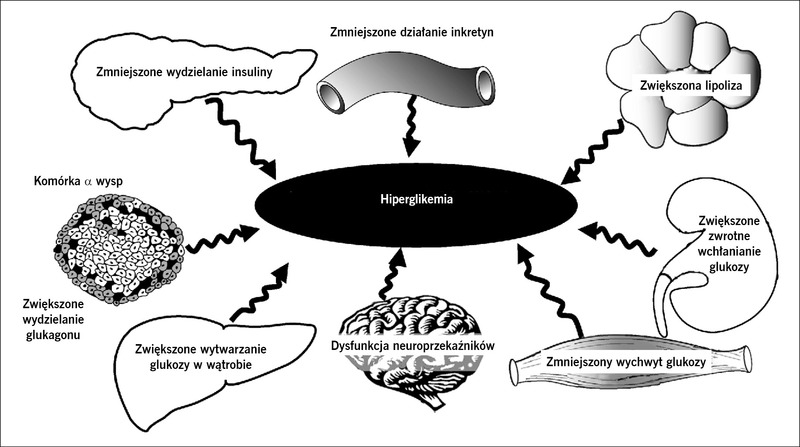
Rycina 1. Złowieszczy oktet. Do rozwoju nietolerancji glukozy w cukrzycy typu 2 przyczyniają się liczne zaburzenia.
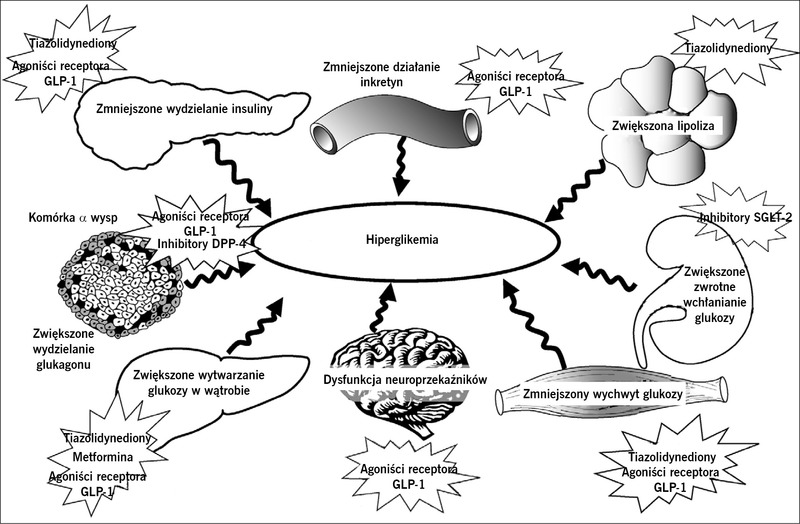
Rycina 2. Zaburzenia patofizjologiczne, na które wpływają obecnie dostępne leki przeciwcukrzycowe.
Od 1987 roku do czasów obecnych nasze rozumienie patofizjologii cukrzycy typu 2 rozszerzyło się od triumwiratu zaburzeń dotyczących komórek β, mięśni i wątroby1 do „złowieszczego oktetu” opisanego w 2008 roku w wykładzie Bantinga2 (ryc. 1). Dowiedzieliśmy się, że niewydolność komórek β występuje znacznie wcześniej w naturalnym przebiegu cukrzycy typu 2, niż sądzono uprzednio, a coraz więcej danych wskazuje na to, że interwencje terapeutyczne, które spowalniają lub opóźniają niewydolność komórek β, mogą prowadzić do trwalszej kontroli glikemii. Celem działania obecnie dostępnych leków przeciwcukrzycowych są liczne defekty patofizjologiczne występujące w cukrzycy typu 2 (ryc. 2), ale kontrola glikemii u chorych na cukrzycę typu 2 pozostaje niedostateczna, ponieważ u około 50% takich osób w Stanach Zjednoczonych stężenie hemoglobiny A1C (HbA1C) przekracza 7,0%. W tym artykule dokonamy przeglądu nowych metod leczenia w zależności od patofizjologii cukrzycy typu 2. W celu przedstawienia potencjalnych celów działania przyszłych leków omówimy również w skrócie patogenezę cukrzycy typu 2.
Czynność komórek β
Podstawowymi zaburzeniami odpowiedzialnymi za cukrzycę typu 2 są upośledzone wydzielanie insuliny spowodowane pogarszającą się czynnością komórek β, zmniejszony wychwyt glukozy przez tkanki obwodowe (mięśnie) oraz zwiększone wytwarzanie glukozy w wątrobie wtórne do wzrostu glukoneogenezy.1,2 Wcześnie w przebiegu choroby wydzielanie insuliny jest zwiększone, ponieważ trzustka próbuje skompensować wzrost stężenia glukozy w osoczu na czczo wynikający z insulinooporności. W miarę dalszego wzrostu stężenia glukozy w osoczu na czczo komórki β nie są jednak w stanie utrzymać zwiększonego tempa wydzielania insuliny i kiedy to wydzielanie zacznie się zmniejszać, rozwija się upośledzona tolerancja glukozy (impaired glukose tolerance, IGT), a w końcu jawna cukrzyca.3-6 Zwiększone wytwarzanie glukozy w wątrobie i zmniejszony wychwyt glukozy przez mięśnie przyczyniają się dodatkowo do stanu hiperglikemii,7,8 który stwarza dalsze obciążenie dla komórek β i zamyka pętlę ujemnego sprzężenia zwrotnego, w którym dekompensacja metaboliczna – glukotoksyczność9 i lipotoksyczność10 – przyczynia się do niewydolności komórek β i nasilenia insulinooporności.
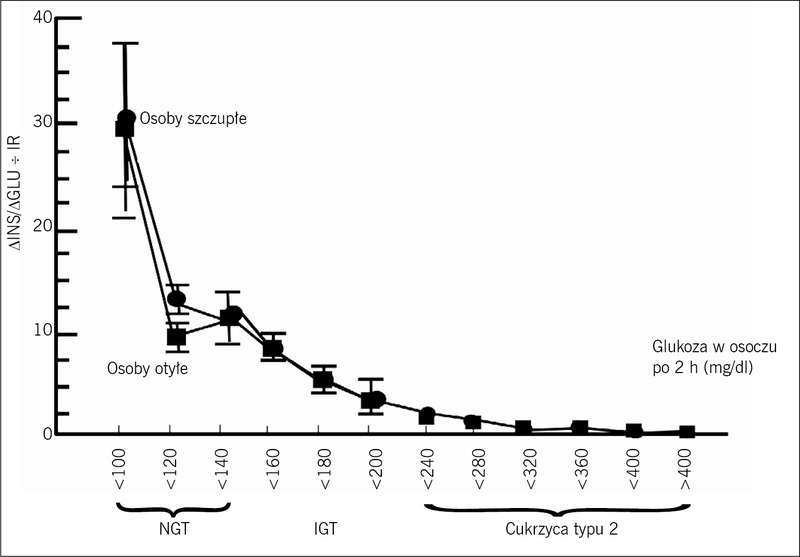
Rycina 3. Wskaźnik wydzielania insuliny/ insulinooporności (wskaźnik podatności na działanie insuliny, disposition index) (ΔINS/ΔGLU ÷ IR) u osób z prawidłową tolerancją glukozy (NGT), upośledzoną tolerancją glukozy (IGT) i cukrzycą typu 2 w funkcji stężenia glukozy w osoczu po 2-godzinnym teście doustnego obciążenia glukozą (szczegółowe omówienie – patrz tekst).
Co szczególnie ważne, odpowiedź insuliny w osoczu na glukozę nie dostarcza informacji na temat stanu zdrowia komórek β. Komórki β odpowiadają na wzrost stężenia glukozy w osoczu wzrostem stężenia insuliny w osoczu, a na tę pętlę sprzężenia zwrotnego wpływa nasilenie insulinooporności. Dlatego też czynność komórek β można najlepiej scharakteryzować za pomocą wskaźnika wydzielania insuliny/insulinooporności (wskaźnik podatności na działanie insuliny, disposition index: ΔINS/ΔGLU ÷ IR, gdzie INS=insulina, GLU=glukoza, IR=insulinooporność mierzona metodą klamry insulinowej).4,11,12 Badania przeprowadzone przez naszą grupę3-5 wykazały, że do niewydolności komórek β dochodzi wcześnie w naturalnym przebiegu cukrzycy typu 2 i jest ona bardziej nasilona, niż pierwotnie sądzono (ryc. 3). Kiedy stężenie glukozy w osoczu po 2-godzinnym teście doustnego obciążenia glukozą (OGTT) u osób z prawidłową tolerancją glukozy zwiększa się od wartości w przedziale od <100 do 100-119 mg/dl do 120-139 mg/dl, czynność komórek β jest już zmniejszona o około 60%. W górnym tercylu IGT (stężenie glukozy w osoczu po 2-godzinnym OGTT=180-199 mg/dl) czynność komórek β jest zmniejszona o 75-80%.4,5,11,12 Bardziej niepokojące niż pogorszenie czynności komórek β jest postępujące zmniejszanie się masy komórek β, które zaczyna się już w stadium przedcukrzycowym i postępuje w miarę pogarszania się cukrzycy. Strategie leczenia chorych na cukrzycę typu 2 powinny więc obejmować stosowanie leków, które opóźniają apoptozę komórek β lub jej zapobiegają.13
W momencie osiągnięcia górnego tercyla IGT większość osób wykazuje już maksymalną lub prawie maksymalną insulinooporność i utraciła większość (75-80%) czynności komórek β. Strategie leczenia chorych na cukrzycę typu 2 powinny więc obejmować stosowanie leków, które podtrzymują czynność komórek β, i charakteryzować się potencjałem prewencji lub opóźniania apoptozy komórek β.
Insulinooporność a cukrzyca typu 2
Insulinooporność, będąca jedną z głównych patofizjologicznych cech cukrzycy typu 2, występuje wcześnie w naturalnym przebiegu tej choroby.1,2,4,8,11,14 Zarówno wątroba, jak i mięśnie wykazują znaczną oporność na działanie insuliny. W cukrzycy typu 2 istnieje ścisła korelacja między wzrostem wytwarzania glukozy w wątrobie a wzrostem stężenia glukozy w osoczu na czczo.1,2 Do zwiększenia szybkości wytwarzania glukozy w wątrobie dochodzi w sytuacji, w której stężenie insuliny w osoczu jest zwiększone dwu-trzykrotnie, co wskazuje na ciężką oporność na działanie insuliny hamujące wytwarzanie glukozy w wątrobie. Kiedy podaje się insulinę we wlewie, uzyskując jej stężenie obserwowane po spożyciu standardowego posiłku, hamowanie wytwarzania glukozy w wątrobie jest znacznie upośledzone.15 Cały wzrost wytwarzania glukozy w wątrobie jest wtórny do przyspieszenia tempa glukoneogenezy wątrobowej.16
Badania, w których stosowano referencyjną metodę normoglikemicznej klamry insulinowej, wykazały, że mięśnie szkieletowe charakteryzuje znaczna insulinooporność, która odpowiada za 85-90% łącznego upośledzenia zużycia glukozy w organizmie u chorych na cukrzycę typu 2.2,8,14,18 Do insulinooporności przyczyniają się liczne wewnątrzkomórkowe zaburzenia jej działania, w tym zmniejszenie transportu i fosforylacji glukozy, zmniejszenie syntezy glikogenu oraz upośledzenie glikolizy i utleniania glukozy.2,19 Co najważniejsze, badania przeprowadzone w naszym laboratorium wykazały, że bardziej proksymalne zaburzenia w obrębie kaskady transdukcji sygnału z receptora insuliny odgrywają ważną rolę w oporności mięśni na insulinę obserwowanej w cukrzycy typu 2.2,11,14,20,21 Należy jednak zauważyć, że chociaż dobrze wykazano insulinooporność w wątrobie i mięśniach we wczesnej fazie choroby, jawna hiperglikemia i cukrzyca nie rozwijają się bez wystąpienia postępującej niewydolności komórek β.1,2,12
Od triumwiratu do złowieszczego oktetu
Oprócz triumwiratu niewydolności komórek β oraz insulinooporności w mięśniach i wątrobie ważną rolę w patogenezie cukrzycy typu 2 odgrywają również inne narządy i tkanki, w tym adipocyty (przyspieszenie lipolizy), przewód pokarmowy (niedobór inkretyn/oporność na inkretyny), komórki α trzustki (hiperglukagonemia), nerki (zwiększenie zwrotnego wchłaniania glukozy/progu nerkowego dla glukozy) oraz ośrodkowy układ nerwowy (insulinooporność). Te liczne zaburzenia określono mianem „złowieszczego oktetu” (ryc. 1).2
Nieharmonijny kwartet
W patogenezie nietolerancji glukozy w cukrzycy typu 2 ważną rolę odgrywają zaburzony metabolizm adipocytów i zmieniona topografia tkanki tłuszczowej.2,10 Komórki tłuszczowe są oporne na hamujące lipolizę działanie insuliny, co prowadzi do zwiększenia stężenia wolnych kwasów tłuszczowych w osoczu15 oraz wzrostu wewnątrzkomórkowego stężenia toksycznych metabolitów lipidów (reszty kwasów tłuszczowych połączone z koenzymem A [fatty acylo-CoA, FACoA], diacyloglicerol [DAG] i ceramidy), co wywołuje insulinooporność w mięśniach i wątrobie22 oraz sprzyja niewydolności komórek β.23 Komórki tłuszczowe są w stanie przewlekłego zapalenia i wydzielają nadmierne ilości cytokin sprzyjających insulinooporności, zapaleniu i miażdżycy (czynnik martwicy nowotworów typu α, interleukina 6, rezystyna i angiotensynogen), natomiast nie wydzielają prawidłowych ilości adipocytokin uwrażliwiających tkanki na działanie insuliny (adiponektyna).10
Katastrofalny kwintet
U chorych na cukrzycę typu 2 obserwuje się również ograniczone działanie inkretyn.2,24,25 Za 90% działania inkretyn odpowiadają dwa hormony inkretynowe, peptyd glukagonopodobny typu 1 (GLP-1) i polipeptyd insulinotropowy zależny od glukozy (GIP), które odgrywają zasadniczą rolę w utrzymywaniu prawidłowej homeostazy glukozy. Zarówno GLP-1, jak i GIP zwiększają wydzielanie insuliny, a GLP-1 hamuje również wydzielanie glukagonu, opóźnia opróżnianie żołądka i zmniejsza łaknienie.26 Mimo że w niektórych badaniach wykazano niewielki defekt wydzielania GLP-1 w cukrzycy typu 2, w innych badaniach udokumentowano prawidłowe lub nawet zwiększone wydzielanie GLP-1.27 Zgodnie obserwuje się natomiast ciężką oporność na pobudzający wpływ GLP-1 na wydzielanie insuliny przez komórki β.28
Szwankujący sekstet
Zwiększone wydzielanie glukagonu przez komórki α i zwiększona wrażliwość wątroby na działanie glukagonu również odgrywają ważną rolę w patofizjologii cukrzycy typu 2.29,30 Glukagon jest czynnikiem o zasadniczym znaczeniu dla utrzymywania zwiększonego podstawowego wytwarzania glukozy w wątrobie u chorych na cukrzycę typu 2.29,30 W porównaniu z osobami z prawidłową tolerancją glukozy stężenie glukagonu w osoczu u osób z IGT i cukrzycą typu 2 jest zwiększone, mimo że występuje u nich hiperglikemia i hiperinsulinemia, które powinny hamować wydzielanie glukagonu. Zwiększone stężenie glukagonu pobudza wytwarzanie glukozy w wątrobie i hamuje działanie insuliny, która z kolei hamuje wytwarzanie glukozy w wątrobie. Obserwuje się również zwiększoną wrażliwość na pobudzający wpływ glukagonu na wytwarzanie glukozy w wątrobie.30 W cukrzycy typu 2, kiedy wydzielanie glukagonu jest hamowane przez somatostatynę, stężenie glukagonu w osoczu na czczo zmniejsza się wraz ze znacznym zmniejszeniem podstawowego wytwarzania glukozy w wątrobie.29