



Spis treści
Choroba Gauchera to wrodzone schorzenie powodowane przez mutację genu GBA1 znajdującego się na chromosomie 1. Jest najczęściej występującą chorobą spichrzeniową.
Częstość występowania w populacji ogólnej wynosi od 1:20 000 do 1:75 000 urodzeń. Zwiększona częstość choroby jest stwierdzana w niektórych grupach etnicznych, np. w Izraelu. W populacji Żydów aszkenazyjskich chorobowość wynosi 1:850 osób.
Etiopatogeneza
Choroba jest dziedziczona w sposób autosomalny recesywny. Stwierdzono wiele różnych
mutacji w genie GBA1, które mogą doprowadzać do jej rozwoju. Rodzaj mutacji może w pewnym stopniu korelować z przebiegiem choroby. Mutacja genu GBA1 doprowadza do nieprawidłowej budowy, niedoboru lub całkowitego braku enzymu lizosomalnego kwaśnej β-glikozydazy (glukocerebrozydazy). W wyniku defektu enzymu dochodzi do zaburzenia metabolizmu fosfolipidów wchodzących w skład błony komórkowej i, co za tym idzie, do gromadzenia się nieprawidłowych metabolitów w makrofagach.
Makrofagi przeładowane glikosfingolipidami w różnych narządach określane są jako komórki Gauchera i znajdują się w dużej liczbie przede wszystkim w śledzionie, wątrobie czy szpiku kostnym. Nagromadzenie się makrofagów obładowanych nieprawidłowymi metabolitami sfingolipidów prowadzi do znacznego powiększenia narządów (śledziony, wątroby) i nieprawidłowego metabolizmu kostnego, osteopenii, z czego wynika zwiększone ryzyko patologicznych złamań, w tym kompresyjnych złamań kręgów. Może dochodzić również do uszkodzenia układu nerwowego.
Typy choroby
Wyróżniamy 3 główne typy choroby: I, II oraz III. W typie I, spotykanym najczęściej, stwierdzamy objawy gromadzenia substancji (pochodnych sfingolipidów), które nie mogą być prawidłowo metabolizowane w związku z niedoborem enzymu β-glukocerebrozydazy lub jego brakiem. Dochodzi więc do gromadzenia nieprawidłowych metabolitów w makrofagach. Obładowane makrofagi doprowadzają do powiększenia śledziony, w mniejszym stopniu wątroby. Mogą również gromadzić się w szpiku kostnym. U pacjentów nierzadko występuje niedokrwistość o złożonej etiologii, jak również małopłytkowość. Dodatkowo często występują dolegliwości bólowe kostne, łącznie z przełomami bólowymi o bardzo dużym nasileniu. Wymienione wyżej objawy występujące w typie I choroby Gauchera sugerują rozrostową chorobę hematologiczną. Z tego powodu dużą wagę przykłada się do podniesienia świadomości dotyczącej symptomatologii tego rzadkiego schorzenia wśród hematologów, ale również lekarzy innych specjalności. Często diagnostyka przed ustaleniem ostatecznego rozpoznania jest długotrwała, a pacjenci wędrują od jednego specjalisty do kolejnego.
Przebieg choroby może być zróżnicowany – od bardzo ciężkiego, z objawami obecnymi już w okresie dzieciństwa, do łagodniejszych postaci. Nierzadko schorzenie to przy skąpoobjawowym przebiegu jest rozpoznawane dopiero u osób dorosłych. W polskiej populacji najczęściej mamy do czynienia z typem I choroby, w którym objawy neurologiczne nie są dominujące.
Choroba może przebiegać w postaci łagodnej, umiarkowanej lub ciężkiej. Wśród objawów występują: hepatosplenomegalia, dolegliwości kostne (ból, martwica kości, złamania patologiczne), tendencja do anemizacji i małopłytkowości. Etiologia cytopenii jest złożona. Pod uwagę bierze się wyparcie prawidłowego utkania krwiotwórczego szpiku przez komórki Gauchera. Do niedokrwistości przyczynia się również niedobór żelaza oraz witaminy B12. Obładowane nieprawidłowymi metabolitami makrofagi nie funkcjonują prawidłowo, zaburzona jest produkcja cytokin, co doprowadza do przewlekłego stanu zapalnego i dysregulacji układu immunologicznego. Z tego powodu u pacjentów z chorobą Gauchera stwierdza się również zaburzenia metabolizmu żelaza typowe dla niedokrwistości chorób przewlekłych. Mogą się także pojawić niedokrwistość i małopłytkowość w mechanizmie autoimmunologicznym. Dysfunkcja immunologiczna powoduje też zwiększoną częstość chorób nowotworowych. U pacjentów z typem I choroby ryzyko rozwoju nowotworu jest 1,7 razy większe w porównaniu z populacją zdrową. Szczególnie wysokie ryzyko dotyczy rozwoju przewlekłych rozrostów limfoproliferacyjnych (4-12 razy większe ryzyko niż w populacji ogólnej), w tym szpiczaka plazmocytowego i innych gammapatii monoklonalnych (ryzyko 25 razy większe niż w populacji ogólnej). Ponadto u pacjentów z chorobą Gauchera występuje zwiększone ryzyko powikłań krwotocznych. Mogą one być związane z małopłytkowością oraz dodatkowo z zaburzeniami koagulologicznymi wynikającymi z upośledzonej produkcji czynników krzepnięcia. Tego typu nieprawidłowości funkcji wątroby pojawiają się w przypadku obecności licznych komórek Gauchera.
Objawy choroby mogą wystąpić w każdym momencie życia – od dzieciństwa po dorosłość. W wieku dorosłym najbardziej dotkliwe są problemy kostne. Pojawiają się przewlekłe, dokuczliwe bóle, niekiedy przyjmują postać ostrych kryz bólowych opornych na standardowe leczenie przeciwbólowe. U dzieci dochodzi do spowolnienia lub zahamowania tempa wzrastania oraz opóźnienia dojrzewania.
Typ II to ciężka postać z dominującymi i wcześnie rozwijającymi się objawami neurologicznymi. Czas przeżycia pacjentów z typem II wynosi około 2 lat. Nie ma skutecznego leczenia w tej postaci choroby.
Typ III (neuropatyczny) charakteryzuje się uszkodzeniem ośrodkowego układu nerwowego (OUN), ale objawy neurologiczne postępują wolniej niż w typie II. Pojawiają się postępująca encefalopatia, różnorodne objawy neurologiczne, które towarzyszą objawom ogólnoustrojowym, podobnym jak w typie I choroby.
Cechy charakterystyczne poszczególnych typów choroby przedstawiono w tabeli 1.
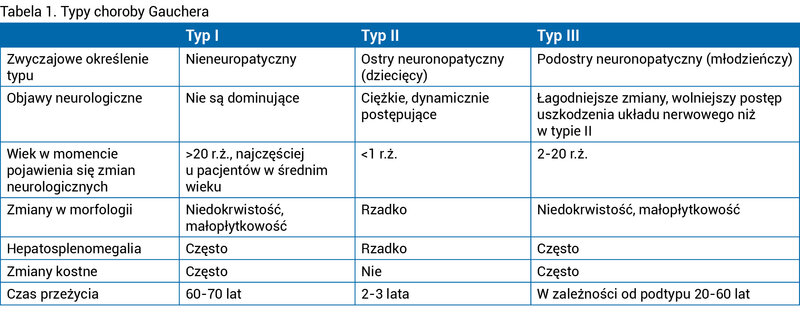
Tabela 1. Typy choroby Gauchera
Diagnostyka
Podstawą rozpoznania jest stwierdzenie braku enzymu β-glukocerebrozydazy lub znacznie obniżonej jego aktywności w leukocytach lub fibroblastach skóry, potwierdzone w badaniu molekularnym. Podejrzenie choroby można wysunąć na podstawie badania histopatologicznego zajętego narządu: szpiku, wątroby czy śledziony, w którym obserwuje się nieprawidłowe makrofagi o piankowatej cytoplazmie. Obecność komórek podobnych do komórek Gauchera (tzw. komórki pseudo-Gauchera) zdarza się w przypadku niektórych chorób hematologicznych, takich jak: chłoniaki nieziarnicze, chłoniak Hodgkina i przewlekła białaczka limfocytowa. Ocena histopatologiczna zajętej tkanki nie jest rekomendowanym badaniem w przypadku podejrzenia choroby Gauchera.
Jak już wspomniano, w diagnostyce główną rolę odgrywają ocena aktywności enzymu β-glukocerebrozydazy oraz badania molekularne, które mogą być przeprowadzone z krwi. Obecnie dostępne są testy bibułowe oparte na ocenie kropli krwi. Badanie jest nieinwazyjne, łatwe do wykonania, a pobrany materiał jest stabilny podczas transportu. Tak pobrany materiał po starannym wysuszeniu może być w łatwy sposób przekazany do laboratorium. Badania w kierunku choroby Gauchera powinno się wykonywać u pacjentów z hepatosplenomegalią, anemizacją, małopłytkowością czy bólami kości o niewyjaśnionej przyczynie. W takim przypadku zazwyczaj wykonywane jest badanie cytologiczne lub histopatologiczne szpiku kostnego. Jest to standardowe postępowanie diagnostyczne zmierzające do wykrycia choroby rozrostowej układu krwiotwórczego, co sugerują powyższe dolegliwości. Dopiero po wykluczeniu chorób hematologicznych jako przyczyny opisywanych odchyleń zlecamy badanie w kierunku choroby Gauchera. Badanie to powinno być wykonywane również u pacjentów przed planowaną splenektomią diagnostyczną w przypadkach izolowanej splenomegalii o niejasnej przyczynie.
Badania określające zaawansowanie choroby
W celu określenia zaawansowania choroby wykonywane są badania biochemiczne, przede wszystkim oceniające funkcje wątroby, morfologia i koagulogram. U pacjentów często stwierdzane są niedobory witaminy B12, nieprawidłowa jest również gospodarka żelazem. W surowicy pacjentów niejednokrotnie wykrywa się obecność białka monoklonalnego. W takim przypadku należy poszerzyć panel badań w kierunku współistniejącej gammapatii monoklonalnej. W ramach diagnostyki obrazowej należy wykonać ultrasonografię (USG) jamy brzusznej z określeniem wielkości wątroby i śledziony, badanie densytometryczne kości (DEXA – dual-energy X-ray absorptiometry) oraz rezonans magnetyczny (MR – magnetic resonance) kości długich (badanie wskazane u pacjentów z nieprawidłowościami układu kostno-stawowego lub w przypadku pojawienia się dolegliwości bólowych).
Leczenie
ERT
W chorobie Gauchera od wielu lat stosowane jest leczenie suplementujące enzym (ERT – enzyme replacement therapy), skuteczne przede wszystkim u pacjentów z typem I choroby. Leczenie to może być również zlecane w typie III, nie hamuje jednak postępu zmian w układzie nerwowym. Wczesne rozpoczęcie terapii substytucyjnej u pacjentów z objawową postacią choroby doprowadza do ustąpienia organomegalii, zmniejszenia dolegliwości kostnych oraz poprawy parametrów morfologicznych, przede wszystkim liczby płytek i stężenia hemoglobiny. Poprawa morfologii i ustąpienie hepatosplenomegalii obserwowane są zazwyczaj po kilku–kilkunastu miesiącach regularnego leczenia. U pacjentów w trakcie terapii substytucyjnej stwierdzano ustąpienie dolegliwości bólowych ze strony kości i zmniejszenie częstości występowania przełomów bólowych. Leczenie zapobiega też pojawieniu się jałowej martwicy kości. Po wielu latach terapii możliwe są również poprawa uwapnienia kości czy ustąpienie osteopenii. Odpowiedź w zakresie dolegliwości ze strony układu kostno-szkieletowego uzyskuje się zazwyczaj później niż poprawę morfologii i redukcję hepatosplenomegalii. Leczenie substytucyjne jest stosowane w formie regularnych, podawanych co 2 tygodnie wlewów dożylnych. Terapia jest zarejestrowana dla pacjentów z typami I (dot. imiglucerazy i welaglucerazy α) oraz III (dot. imiglucerazy) choroby, u których występują objawy. Leczenie nie jest wskazane u osób z asymptomatyczną (bezobjawową) postacią choroby Gauchera. Dawka leku zależy od ciężkości objawów i wynosi 15-60 j./kg m.c. W Unii Europejskiej obecnie są zarejestrowane dwa preparaty ERT: imigluceraza i welagluceraza α.
SRT
Inną opcją jest zastosowanie terapii redukującej substrat (SRT – substrate reduction therapy). Leki działające poprzez hamowanie syntazy glukozyloceramidu mają formę doustną. W Unii Europejskiej jest zarejestrowany eliglustat. Trzeba pamiętać, że lek ten jest metabolizowany w wątrobie przez cytochrom p450, przede wszystkim przez jego izoenzym CYP2D6. Należy poinformować pacjenta o możliwych interakcjach z innymi lekami, które mogą hamować lub indukować izoenzym CYP2D6. W przypadku eliglustatu cele terapeutyczne są określone podobnie jak przy stosowaniu ERT.
Inne opcje terapeutyczne
Wobec wysokiej skuteczności stosowanego leczenia obecnie rzadko przeprowadzane są w terapii choroby Gauchera transplantacje allogenicznych komórek macierzystych hematopoezy. Procedurę taką można rozważać w przypadku oporności na ERT i SRT, przede wszystkim u pacjentów z typem III choroby.
Leczenie w Polsce
W Polsce decyzję o kwalifikacji pacjentów z chorobą Gauchera do terapii ERT i SRT podejmuje Zespół Koordynacyjny ds. Chorób Ultrarzadkich na podstawie wniosku lekarza prowadzącego i zgromadzonej dokumentacji medycznej. Leczenie podlega okresowej kontroli i stosowane jest przewlekle w przypadku potwierdzenia jego dobrej skuteczności i tolerancji.
Podsumowanie
Choroba Gauchera to rzadka choroba uwarunkowana genetycznie. Objawy występujące w najczęstszym typie I powodują, że pacjent jest kierowany do specjalistycznej diagnostyki. Zazwyczaj jednak, zanim choroba zostanie zdiagnozowana, wędruje przez gabinety lekarzy różnych specjalności. Należy pomyśleć o tej jednostce chorobowej w przypadku pojawienia się cytopenii we krwi obwodowej, powiększenia śledziony lub wątroby oraz przewlekłych, okresowo nasilających się bólów kostnych. Ustalenie właściwego rozpoznania jest tym bardziej kluczowe, że dysponujemy skuteczną terapią, która zapobiega progresji objawów związanych z kumulacją obładowanych nieprawidłowymi metabolitami makrofagów.
Materiał dla lekarzy, przygotowany z inicjatywy i sponsorowany przez Takeda.
C-APROM/PL/GAUD/0037, 05/2021
© 2021 Takeda Pharmaceutical Company Limited. Wszystkie prawa zastrzeżone. Wszystkie znaki towarowe są własnościami ich prawowitych właścicieli.

Takeda Pharma Sp. z o.o.
ul. Prosta 68, 00-838 Warszawa
tel. +48 22 608 13 00 lub 01, faks: +48 22 608 13 03
www.takeda.com/pl-pl/
Następny artykuł:

 Dodaj do ulubionych
Dodaj do ulubionych
