Rehabilitacja neurologiczna
Czynniki wpływające na zaburzenia chodu u pacjentów z chorobą Charcota-Mariego-Tootha
mgr Katarzyna Bienias1, dr n. med. Joanna Cegielska1, dr n. med. Joanna Ścibek2, dr hab. n. med. Jan Kochanowski1
Dziedziczna neuropatia ruchowo-czuciowa jest najczęstszą neuropatią uwarunkowaną genetycznie. Jest to choroba charakteryzująca się powolnym przebiegiem i dużą różnorodnością objawów, wśród których najbardziej typowe są deformacje stóp, nieprawidłowości postawy i chodu, postępujące symetryczne niedowłady i zaniki mięśniowe w dystalnych odcinkach kończyn oraz zaburzenia czucia. Na początku choroby osłabienie i zaniki mięśniowe zwykle dotyczą mięśni wewnętrznych stopy. Później osłabieniu ulegają pozostałe mięśnie kończyn dolnych, zazwyczaj w następującej kolejności: mięśnie strzałkowe, mięsień prostownik długi palców, mięsień piszczelowy przedni i mięsień trójgłowy łydki. Zmiany w obrębie stopy i stawu skokowo-goleniowego dotyczą aż 94% chorych. Vinci i wsp. opisali kolejne etapy zaburzeń ruchowych w obrębie kończyn dolnych, które dotyczą przede wszystkim odcinków dystalnych. W każdym kolejnym stadium dołącza się nowy objaw, który zmienia wzorzec chodu pacjenta i wymaga różnych interwencji fizjoterapeutycznych. Przedstawiona klasyfikacja bierze pod uwagę zaburzenia spowodowane przez postępujące osłabienie mięśni kończyn dolnych i wynikające z tego problemy biomechaniczne podczas chodu. Podział ten może być pomocny w codziennej praktyce, dając możliwość łatwej oceny zaawansowania dysfunkcji w obrębie kończyn dolnych.
Wprowadzenie
Dziedziczna neuropatia ruchowo-czuciowa (hereditary motor sensory neuropathy, HMSN) należy do złożonej i heterogennej grupy dziedzicznych chorób nerwów obwodowych. Znana jest również pod nazwą choroby Charcota-Mariego-Tootha (CMT) i jest najczęstszą neuropatią uwarunkowaną genetycznie.1-4
Na podstawie badań elektrofizjologicznych HMSN można podzielić na dwie główne grupy, przyjmując za kryterium podziału szybkość przewodzenia nerwowego we włóknach ruchowych nerwu pośrodkowego. Do pierwszej grupy (CMT1) zalicza się postać demielinizacyjną, gdzie występuje znaczne zwolnienie prędkości przewodzenia nerwowego (<38 m/s). Druga grupa to postać aksonalna (CMT2) z prawidłową prędkością przewodzenia lub tylko nieznacznie obniżoną.1-4 W większości przypadków choroba CMT dziedziczona jest w sposób autosomalny dominujący. Najczęstszą mutacją odpowiadającą za wystąpienie choroby w dziedzicznych neuropatiach ruchowo-czuciowych jest duplikacja w regionie 17p11.2-p12, która swoim zasięgiem obejmuje gen kodujący białko mieliny obwodowej. Jest ona odpowiedzialna aż za około 70-80% przypadków CMT1.1-7
Choroba Charcota-Mariego-Tootha należy do wolno postępujących chorób nerwowo-mięśniowych i charakteryzuje się dużą różnorodnością objawów. Typowe symptomy występujące u pacjentów z dziedziczną neuropatią to: deformacje stóp, nieprawidłowości postawy i chodu, postępujące symetryczne niedowłady i zaniki mięśniowe w dystalnych odcinkach kończyn oraz zaburzenia czucia (głównie wibracji). Zaburzenia czucia zwykle nie są przez pacjentów odczuwane mimo że włókna czuciowe są uszkodzone.1-7 Główną konsekwencją neuropatii jest osłabienie siły mięśniowej (atrofia mięśni na skutek odnerwienia wtórnego lub pierwotnego w zależności od postaci).
Ostatnie badania pokazały, że w porównaniu z osobami zdrowymi u pacjentów z CMT siła mięśniowa jest obniżona we wszystkich mięśniach, jednak za mięsień rzeczywiście osłabiony należy uznać ten, który jest niezdolny do pełnej aktywności funkcjonalnej, do której jest przeznaczony. W związku z tym w pierwszych stadiach choroby mięsień trójgłowy łydki pozostaje silny, ponieważ spełnia swoje zadanie funkcjonalne podczas chodu, mimo że zazwyczaj w badaniu elektrofizjologicznym obserwuje się zmiany związane z odnerwieniem już w momencie wystąpienia objawów klinicznych.8
Na początku choroby osłabienie i zaniki mięśniowe zwykle dotyczą mięśni wewnętrznych stopy. Dolegliwości w obrębie stopy i stawu skokowo-goleniowego dotyczą aż 94% chorych.9 Później osłabieniu ulegają kolejne mięśnie kończyn dolnych, zazwyczaj w następującej kolejności: mięśnie strzałkowe (nawracające stopę), mięsień prostownik długi palców i palucha, mięsień piszczelowy przedni (zginacze grzbietowe stawu skokowo-goleniowego) i ostatecznie mięsień trójgłowy łydki (zginacz podeszwowy stawu skokowo-goleniowego).8 W zaawansowanym stadium choroby może dochodzić do osłabienia siły mięśni znajdujących się powyżej stawów kolanowych.5,8,10
Ponieważ obecnie niedostępna jest ani terapia lekowa ani genowa CMT, jedynym sposobem leczenia, dzięki któremu realna jest poprawa możliwości funkcjonalnych oraz jakości życia pacjentów jest fizjoterapia.8,10 Dla lekarzy i fizjoterapeutów ważna jest wiedza na temat typowego przebiegu choroby, ponieważ cechą charakterystyczną CMT jest nierównomierny rozkład osłabienia siły mięśniowej, gdzie niektóre grupy mięśniowe są bardziej dotknięte procesem chorobowym niż inne. Vinci i wsp. opisali kolejne etapy zaburzeń ruchowych w obrębie kończyn dolnych, które dotyczą przede wszystkim odcinków dystalnych. Ten funkcjonalny podział bierze pod uwagę zaburzenia spowodowane postępującym osłabieniem mięśni kończyn dolnych i wynikające z tego problemy biomechaniczne podczas chodu. W każdym kolejnym stadium dołącza się nowy objaw, który zmienia wzorzec chodu pacjenta oraz wymaga różnych interwencji fizjoterapeutycznych.8
Rozwój zaburzeń mięśniowych w obrębie kończyn dolnych
Opadanie przodostopia
W przebiegu CMT jako pierwsze osłabieniu ulegają mięśnie wewnętrzne stopy: zginacz palucha krótki, mięśnie glistowate i mięśnie międzykostne. Te drobne mięśnie fizjologicznie stabilizują stawy śródstopno-paliczkowe, tj. utrzymują palce w wyproście podczas gdy w fazie przenoszenia kończyny dolnej mięsień prostownik długi palucha i prostownik długi palców kurczą się, aby unieść przodostopie. Gdy brakuje stabilizacji dla stawów śródstopno-paliczkowych, paliczek bliższy każdego palca ustawia się w nadmiernym wyproście. Jest on pociągany przez prostownik długi palucha i prostownik długi palców i prześlizguje się po głowie kości śródstopia w kierunku grzbietowym, a głowa kości śródstopia ulega obniżeniu (ryc. 1).8,10,11 Dochodzi do ustawienia przodostopia w zgięciu podeszwowym, a kąt między osią kończyny dolnej a styczną do podeszwy stopy zwiększa się (w fazie przenoszenia powinien wynosić 90 stopni), ale na tym etapie nie przekracza 100 stopni. Kończyna dolna staje się funkcjonalnie dłuższa. Zwiększone zgięcie podeszwowe pierwszego promienia stopy powoduje powiększenie wysokości łuku stopy, co prowadzi do powstania stopy wydrążonej, będącej głównym objawem choroby CMT.8-10,12
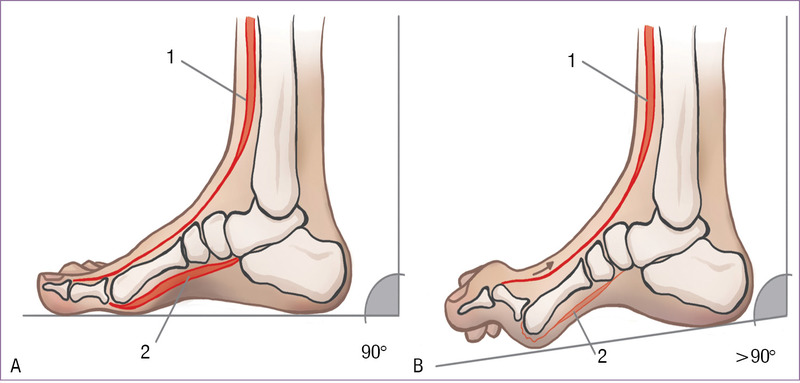
Rycina 1. Schematyczne przedstawienie ustawienia stopy u osoby zdrowej (A) oraz u chorego z HMSN (B). Na schemacie (B) widoczne obniżenie głowy śródstopia oraz zwiększenie kąta >90 stopni między osią kończyny dolnej a styczną do podeszwy stopy. 1 – mięsień prostownik długi palucha i prostownik długi palców, 2 – mięśnie wewnętrzne stopy (zginacz palucha krótki, mięśnie glistowate i międzykostne). Opracowanie własne na podstawie:8,10
Pacjenci starają się kompensować opadanie przodostopia przez silniejsze skurcze mięśni prostownika długiego palucha i prostownika długiego palców, ale brak stabilizacji stawów śródstopno-paliczkowych prowadzi tylko do pogłębienia dysfunkcji. Inną metodą samodzielnej kompensacji jest ustawienie stawu skokowo-goleniowego w zgięciu grzbietowym dzięki silnemu jeszcze na tym etapie mięśniu piszczelowemu przedniemu. Jest to użyteczny mechanizm dla funkcjonalności chodu, ale nadaktywność mięśnia piszczelowego przedniego prowadzi do ustawienia stopy w supinacji i ewolucji zaburzeń w obrębie stopy do kolejnego stadium.8
Ponadto deformacji paliczków bliższych w postaci ich przeprostu towarzyszy zwykle zgięcie w stawach międzypaliczkowych bliższych, co powoduje powstanie palców szponiastych. To nieprawidłowe ustawienie zmniejsza płaszczyznę podparcia, powodując ból i trudności w dobraniu obuwia.5,8-10
W czasie chodu najłatwiejszym dla pacjentów odruchowym sposobem kompensowania opadania przodostopia jest zwiększenie zgięcia w stawie kolanowym podczas fazy przenoszenia kończyny dolnej.8,13 Taka strategia zmniejsza prędkość chodu, ale zachowuje prawidłową długość kroku mimo większego zużycia energii.13
Na tym etapie u pacjentów powinno się rozważyć pewne działania poprawiające funkcjonowanie. Zapobieganie utrwaleniu deformacji w obrębie stawów śródstopno-paliczkowych jest możliwe przez:
- stosowanie indywidualnie dobranych wkładek ortopedycznych wyposażonych w klin pod śródstopie, który w fazie obciążania kończyny dolnej przywraca ustawienie stawów śródstopno-paliczkowych do niewielkiego zgięcia,
- codzienne rozciąganie grzbietowej części torebki stawowej stawów śródstopno-paliczkowych, utrzymując stawy międzypaliczkowe w wyproście.
Zapobieganie przykurczom mięśni zginających stopę podeszwowo i zmniejszeniu zgięcia grzbietowego stawu skokowo-goleniowego jest możliwe przez:
- rozciąganie mięśnia trójgłowego łydki,
- noszenie w domu obuwia z płaską podeszwą (funkcjonalne rozciąganie ścięgna Achillesa).
Ponadto powinno się dołączyć ćwiczenia poprawiające równowagę. Poprawę funkcjonalności chodu można uzyskać przez noszenie butów z niewielkim obcasem (do 3 cm), które niwelują potrzebę zgięcia grzbietowego stawu skokowo-goleniowego powyżej 90 stopni. W kolejnych etapach rozwoju zaburzeń w obrębie kończyn dolnych powinno się kontynuować wymienione działania, dołączając na każdym etapie postępowanie nakierowane na pojawiające się nowe objawy.8