Wynik badania elektromiograficznego jest podobny jak w SMA. Ponadto w SBMA stwierdza się cechy uszkodzenia neurogennego z obecnością licznych fascykulacji w mięśniach regionu opuszki. Przewodnictwo czuciowe jest nieprawidłowe. Rejestruje się obniżone amplitudy odpowiedzi czuciowych z nerwów obwodowych.
SBMA wymaga różnicowania przede wszystkim z ALS. W ALS nie stwierdza się drżenia rąk, objawów czuciowych ani zaburzeń endokrynologicznych. Ponadto postęp choroby w ALS jest dużo szybszy. Rozpoznanie SBMA, podobnie jak SMA, ustalane jest na podstawie badania genetycznego.
Dziedziczne neuropatie ruchowe
Dziedziczne neuropatie ruchowe (dHMN – distal hereditary motor neuropathies) to bardzo rzadka i niezwykle heterogenna pod względem genetycznym i klinicznym grupa chorób dolnego neuronu ruchowego, które mogą być dziedziczone w sposób autosomalnie dominujący, autosomalnie recesywny lub sprzężony z chromosomem X. Dotychczas odkryto mutację kilkunastu genów odpowiedzialnych za rozwój choroby. Mutacja jednego genu może powodować różne fenotypy choroby. Szacuje się jednak, że w 80% przypadków dHMN są spowodowane mutacją jeszcze nieodkrytego genu. U większości chorych objawy rozwijają się w ciągu dwóch pierwszych dekad życia (tab. 5).
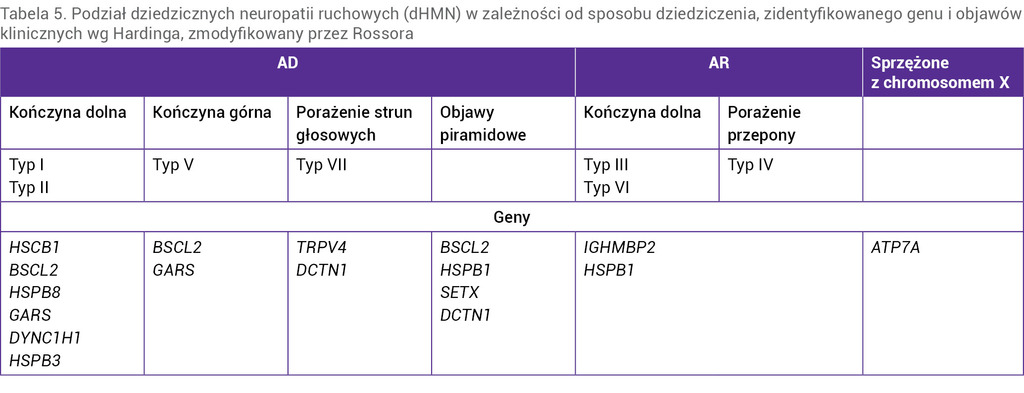
Tabela 5. Podział dziedzicznych neuropatii ruchowych (dHMN) w zależności od sposobu dziedziczenia, zidentyfikowanego genu i objawów klinicznych wg Hardinga, zmodyfikowany przez Rossora
Dziedziczne neuropatie ruchowe charakteryzują się powoli postępującym, symetrycznym i głównie dystalnym niedowładem kończyn dolnych, bez towarzyszących zaburzeń czucia. W dHMN nie występują objawy opuszkowe. Do rozwoju objawów dochodzi w dzieciństwie, a początek ich wystąpienia jest trudny do wychwycenia. Objawy zazwyczaj rozpoczynają się w obrębie stóp (stopa wydrążona, płaska, palce młotkowate), z czasem wstępują. Wyjątkiem jest typ V wg Hardinga, w którym pierwsze objawy rozwijają się w obrębie kończyn górnych.
Oprócz niedowładu kończyn w niektórych typach dHMN występują objawy towarzyszące, np. porażenie przepony lub porażenie strun głosowych (tab. 5).
Istotną rolę w diagnostyce dHMN odgrywa badanie elektrofizjologiczne, które pozwala różnicować tę grupę chorób z polineuropatiami ruchowo-czuciowymi uwarunkowanymi genetycznie (CMT – Charcot-Marie-Tooth disease) oraz z dystalnymi miopatiami. W badaniu przewodnictwa nerwowego stwierdza się obniżone amplitudy odpowiedzi ruchowych; zmiany są zależne od długości włókien. W przeciwieństwie do CMT przewodnictwo czuciowe jest prawidłowe. Badanie elektromiograficzne potwierdza przewlekle neurogenne uszkodzenie mięśni dystalnych.
Rozpoznanie choroby ustala się na podstawie obrazu klinicznego, wyniku badania elektrofizjologicznego i wyniku badania genetycznego.
Dziedziczne paraparezy spastyczne
Dziedziczne paraparezy spastyczne (HSP – hereditary spastic paraplegia) to grupa chorób, w przebiegu których dochodzi do uszkodzenia górnego neuronu ruchowego. Stanowią heterogenną grupę schorzeń, które mogą być dziedziczone w sposób autosomalnie dominujący, autosomalnie recesywny, sprzężony z chromosomem X lub mitochondrialnie. Opisywane są również przypadki sporadyczne. Dotychczas odkryto ponad 70 genów odpowiedzialnych za rozwój choroby (tab. 6).
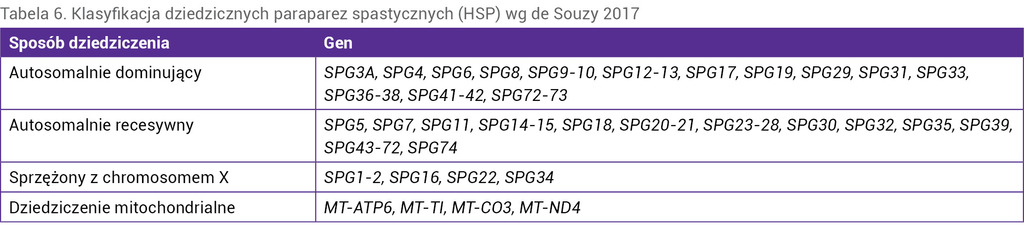
Tabela 6. Klasyfikacja dziedzicznych paraparez spastycznych (HSP) wg de Souzy 2017
HSP występują rzadko. Chorobowość wynosi 1,8/100 000. Choroba postępuje powoli. Paraparezy spastyczne dziedziczone w sposób autosomalnie dominujący rozwijają się zazwyczaj w wieku dorosłym (między drugą a trzecią dekadą życia), a dziedziczone w sposób autosomalnie recesywny – w dzieciństwie lub u młodych dorosłych.
Głównym objawem HSP jest spastyczny niedowład kończyn dolnych, który wynika z uszkodzenia dróg korowo-rdzeniowych. Mogą mu towarzyszyć zaburzenia zwieraczowe lub czucia głębokiego.
HSP dzielimy na formy proste i złożone. W przypadku tych pierwszych niedowład jest jedynym objawem choroby. W przypadku współistniejącego zajęcia innych układów paraparezę spastyczną określamy jako złożoną. W formie złożonej HSP może dochodzić do zajęcia móżdżku, układu pozapiramidowego, nerwów obwodowych lub mięśni. W tej grupie chorych mogą występować także zaburzenia funkcji poznawczych lub psychiczne. Niekiedy u chorych występują cechy dysmorfii (zmiany w obrębie twarzy, niski wzrost), malformacje kostne (skolioza, malformacje w obrębie stóp) lub zajęcie układu wzrokowego (zaćma, zwyrodnienie barwnikowe siatkówki, zanik nerwu wzrokowego). W badaniach obrazowych chorych na HSP złożoną stwierdza się nieprawidłowości takie jak zmiany w obrębie istoty białej mózgu, ścieńczenie ciała modzelowatego, zanik rdzenia kręgowego lub móżdżku.
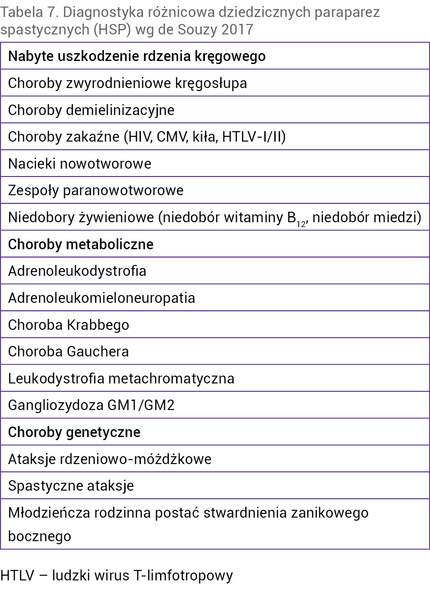
Tabela 7. Diagnostyka różnicowa dziedzicznych paraparez spastycznych (HSP) wg De Souzy 2017
Klasyfikacje HSP uwzględniają sposób dziedziczenia, formę choroby (prosta lub złożona) lub zmutowany gen (SPG – spastic paraplegia genes).