IV edycja Kongresu HPV już 12-13 czerwca! Poznaj najnowsze trendy w profilaktyce, diagnostyce i leczeniu raka szyjki macicy i innych schorzeń związanych z HPV | Sprawdź >
Aktualne osiągnięcia
Genetyczne zróżnicowanie ostrych białaczek limfoblastycznych
Mgr Karolina Karabin, dr n. med. Marta Libura
Celem poniższej pracy jest przedstawienie aktualnych osiągnięć w dziedzinie genetyki OBL u dzieci i dorosłych w świetle ich klinicznego znaczenia oraz roli tych badań na tle innych metod diagnostycznych stosowanych w rutynowej praktyce laboratoryjnej
WPROWADZENIE
Ostre białaczki limfoblastyczne (OBL) są heterogenną grupą chorób nowotworowych dotyczącą niedojrzałych komórek limfoidalnych, które naciekają szpik kostny oraz krew, a także mogą tworzyć nacieki w różnych organach. OBL z różną częstością występują u osób w każdej grupie wiekowej, ale u dzieci stanowią 80 proc. wszystkich białaczek, ze szczytem zachorowania pomiędzy 2. a 5. r.ż. U dorosłych OBL stanowią 20 proc. wszystkich ostrych białaczek, zaś ich występowanie obserwuje się przeważnie przed 30. r.ż..[1] W OBL u dzieci uzyskuje się wyleczenie u 3/4 chorych. Natomiast pomimo dużego postępu w dziedzinie leczenia OBL, u większości dorosłych i niemowląt terapia kończy się niepowodzeniem, co oznacza śmierć pacjenta.[2]
Ten fakt wzmacnia konieczność poznania mechanizmów transformacji OBL, a następnie modyfikacji aktualnego systemu klasyfikacyjnego, tak aby umożliwić podejmowanie prób opracowania nowych form terapii, lepiej dostosowanych do choroby u danej osoby. Terapie te, poprzez ingerencję w mechanizmy prowadzące do rozwoju choroby, byłyby bardziej skuteczne w zabijaniu komórek białaczkowych, a przy tym mniej toksyczne dla komórek zdrowych. Aby ustalić wskazania do takich terapii, niezbędny byłby indywidualny dobór pacjentów, możliwy do przeprowadzenia na podstawie metod diagnostycznych, które obecnie stosowane są wyłącznie w celach badawczych.
DOTYCHCZASOWE SYSTEMY KLASYFIKACYJNE W OBL
Podstawowy podział OBL dokonywany jest na podstawie dominującego w obrazie rodzaju komórki, stąd OBL zostały następnie podzielone na:
- białaczkę B-prekursorową (B-OBL),
- T-prekursorową (T-OBL).[1]
Pierwotny podział B-OBL w ramach klasyfikacji FAB (ang. French-American-British) ma już tylko znaczenie historyczne. Podział EGIL dla T-OBL (ang. European Group for the Immunological Classification of Leukemias), opierający się na wyniku badania immunofenotypowego, określa fizjologiczny etap różnicowania limfocytu, któremu odpowiada dominujący liczbowo rodzaj komórek białaczkowych. Liczne badania potwierdzają jednak, że u podłoża poszczególnych podtypów białaczek, które zostały zdefiniowane na podstawie cytomorfologii oraz immunofenotypu, leżą swoiste zaburzenia genetyczne.
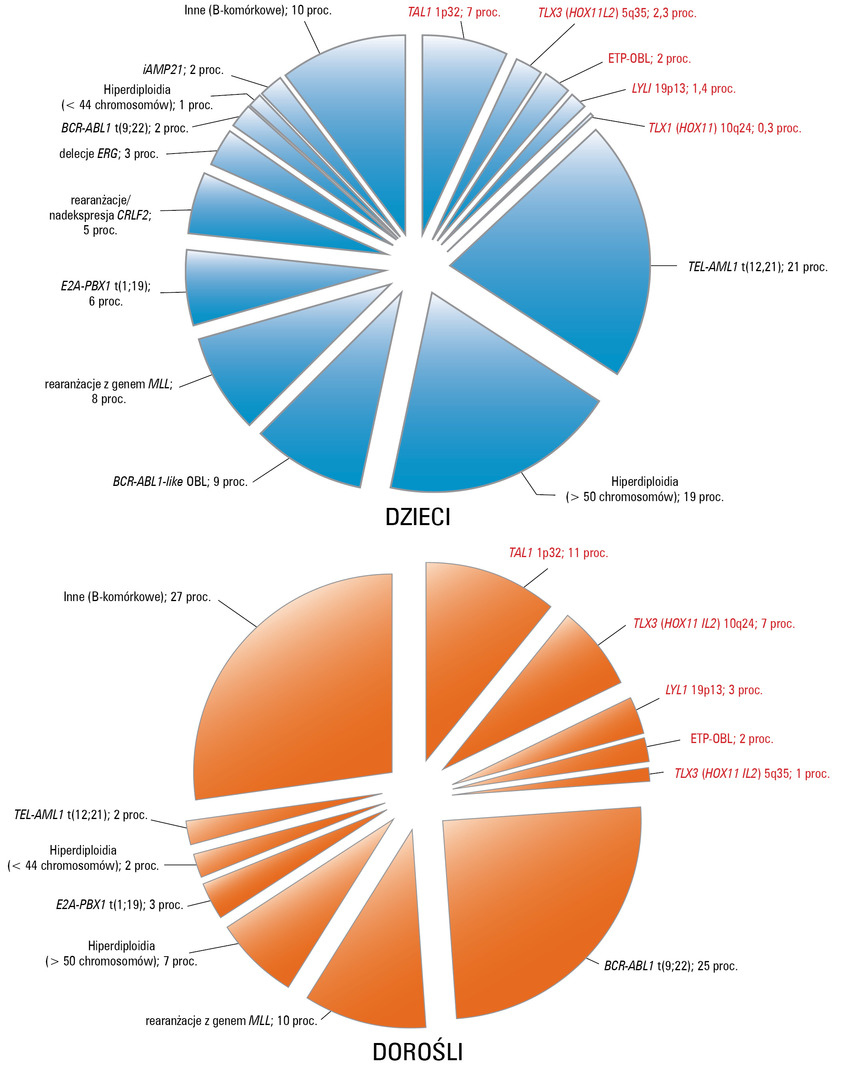
Ryc. 1. Wykresy przedstawiają częstotliwość występowania poszczególnych zmian cytogenetycznych i genetycznych w B-OBL (czarny tekst) i T-OBL (czerwony tekst) u dzieci i dorosłych. Wyjaśnienia skrótów w tekście.
W tej sytuacji kolejne systemy klasyfikacyjne, w tym aktualna wersja WHO 2008,[3] uwzględniają wybrane zmiany genetyczne jako podstawowe kryterium definiowania podjednostek choroby. Dzięki uwzględnieniu zaburzeń cytogenetycznych w kryteriach podziału, klasyfikacja WHO 2008 pozwala w sposób bardziej precyzyjny, niż czyniły to poprzednie wersje FAB i EGIL ustalić ryzyko nawrotu choroby.
Ponieważ klasyfikacja ta bierze pod uwagę jedynie duże zmiany chromosomalne, w założeniu nie jest doskonała, gdyż nie umożliwia subklasyfikacji dużej grupy pacjentów z kariotypem prawidłowym. Dalsza diagnostyka oraz różnicowanie ryzyka nawrotu choroby tej grupy stają się możliwe dzięki zastosowaniu bardziej zaawansowanych metod badań genetycznych, które poddają analizie cały genom: wysokorozdzielcze techniki, oparte głównie na technice mikromacierzy i łańcuchowej reakcji polimerazy (PCR). Za pomocą tych metod, w grupie pacjentów bez dużych aberracji chromosomalnych – w ostatnich kilku latach wykryto obecność wielu zmian na poziomie pojedynczych nukleotydów (tzw. rearanżacje wewnątrzgenowe).[4] Dodatkowo okazuje się również, że zmiany te towarzyszą wielu dużym translokacjom chromosomalnym i ich obecność różnicuje rokowanie w danym cytogenetycznym podtypie OBL. Ostatnie badania wyjaśniają również mechanizmy niestabilności genetycznej w OBL, która prowadzi do rozwoju wtórnych aberracji genetycznych i przyczynia się do fatalnego przebiegu wielu podtypów białaczek.
Dzięki badaniom obejmującym analizę całego genomu, lista nowych aberracji genetycznych w OBL z roku na rok się wydłuża. Stworzenie zatem idealnej klasyfikacji, która uwzględniałaby wszystkie zaburzenia genetyczne i jednocześnie korelowała z klinicznym przebiegiem choroby, wydaje się być niezmiernie trudne. Niezbędne zatem jest całościowe podejście do zagadnienia transformacji OBL, uwzględniające pojawienie się tzw. aberracji pierwotnych, mających kluczowe znaczenie dla rozwoju patologii (ang. driver mutations), jak również dynamikę procesu nabywania mutacji wtórnych, w tym pojawianie się mutacji mniej istotnych dla procesu transformacji (ang. passenger mutations).
GENETYCZNA CHARAKTERYSTYKA B-OBL
Na podstawie klasyfikacji WHO w OBL wywodzącej się z linii B-komórkowej wyróżnia się podklasy cytogenetyczne związane z występowaniem dużych aberracji chromosomalnych: hiperdiploidii (> 50 chromosomów), hipodiploidii (< 50 chromosomów) oraz translokacji: t(9;22)(q34;q11.2); BCR-ABL1, t(12;21);(p13;q22); TEL-AML1, t(1;19); (q23;p13.3); E2A-PBX1, t(5;14)(q31;q32); IL3-IGH, a także rearanżacji genu MLL t(v;11q23).[3]
B-OBL z genem fuzyjnym BCR-ABL1
W wyniku wymiany części ramion długich pomiędzy chromosomem 9 a chromosomem 22 powstaje tzw. chromosom Filadelfia, zawierający gen fuzyjny BCR-ABL1 kodujący białko o aktywności kinazy tyrozynowej. Nadekspresja tego białka jest charakterystyczna dla chorych z przewlekłą białaczką szpikową (PBSz), a także 25 proc. OBL u dorosłych i 2-5 proc. OBL u dzieci.[5]
Klinicznie pacjenci z OBL BCR-ABL1-dodatni są starsi, mają większą liczbę blastów we krwi obwodowej i zaliczani są do grupy wysokiego ryzyka: otrzymują intensywniejszą chemioterapię oraz w miarę możliwości są kwalifikowani do procedur przeszczepienia komórek macierzystych krwi w pierwszej remisji.[6]
Wyniki leczenia istotnie zmieniło wprowadzenie inhibitorów kinaz tyrozynowych (imatynib, dazatynib, nilotynib), dzięki którym rokowanie chorych bardzo się poprawiło. Okazuje się jednak, że grupa pacjentów BCR-ABL1-dodatnich nie jest jednorodna, jeśli chodzi o dynamikę postępu choroby. Badania Cazzaniga i wsp. na bliźniętach jednojajowych, które posiadały taką samą chorobę OBL BCR-ABL1-dodatnią wykazały, że pomimo wspólnej cechy, jaką jest obecność białka BCR-ABL1, kliniczny przebieg choroby może diametralnie różnić się w poszczególnych przypadkach. W badaniu tym jedno z dwójki bliźniąt w każdej z par zmarło wskutek szybkiego nawrotu choroby, mimo zastosowania transplantacji komórek macierzystych, podczas gdy drugie z bliźniąt nie przejawiało tak agresywnego przebiegu choroby. Czynnikiem różnicującym i wpływającym na ewolucję białaczki okazała się obecność mutacji punktowych/delecji genu IKZF1: stwierdzono bowiem, że bliźnięta, które zmarły z powodu nawrotu choroby, posiadały takie mutacje, podczas gdy te, które przeżyły – nie.[7]
Gen IKZF1 koduje bardzo ważny czynnik transkrypcyjny – IKAROS, biorący udział w rozwoju limfocytów B. Delecje IKZF1 często pojawiają się u pacjentów B-OBL BCR-ABL1-dodatnich (ponad 80 proc.).[8] Ostatnie badania wykazują, że są one obecne także u 1/3 pacjentów pediatrycznych B-OBL-negatywnych pod kątem genu BCR-ABL, u których nie stwierdza się także obecności innych genów fuzyjnych (kariotyp prawidłowy).[9] W każdym z przypadków ich obecność związana jest z krótszym całkowitym przeżyciem oraz większym ryzykiem nawrotu choroby.