


Spis treści
- Definicja
- Rys historyczny
- Epidemiologia i zagadnienia genetyczne
- Klasyfikacja leukodystrofii. Rola badań neuroobrazowych
- Budowa istoty białej
- Patogeneza leukodystrofii i genetycznie uwarunkowanych leukoencefalopatii
- Diagnostyka leukodystrofii
- Leukodystrofie hipomielinizacyjne (HLD)
- Wrodzone choroby istoty białej z demielinizacją
- Leukodystrofia metachromatyczna (MLD)
- Leukodystrofia globoidalna Krabbego (GLD)
- Adrenoleukodystrofia sprzężona z chromosomem X (X-ALD)
- Leukodystrofia z zajęciem pnia mózgu i rdzenia oraz podwyższeniem stężenia mleczanów (LBSL)
- Leukodystrofia ze znikającą istotą białą (VWM)
- Choroba Alexandra (ALXDRD)
- Choroba Canavan (CD)
- Leukodystrofia z wielkogłowiem i obecnością torbieli podkorowych (MLC)
- Leukodystrofia demielinizacyjna dominująca o początku w wieku dorosłym (ADLD)
- Leukodystrofia z obecnością sferoidów aksonalnych i pigmentacją gleju (HDLS)
- Genetycznie uwarunkowane leukoencefalopatie (gLE)
- Leczenie i postępowanie we wrodzonych chorobach istoty białej
W pracy omówiono dwie duże grupy leukodystrofii oraz genetycznie uwarunkowane leukoencefalopatie. Leukodystrofie są chorobami bardzo rzadkimi. Z tego względu stanowią duże wyzwanie diagnostyczne dla lekarzy. Dzięki wprowadzeniu badań neuroobrazowych i rozwojowi diagnostyki molekularnej dokonał się ogromny postęp w rozpoznawaniu tych chorób. Wczesne ustalenie rozpoznania przyczynowego pozwala wdrożyć właściwe postępowanie lecznicze, a rodzinę objąć poradnictwem genetycznym.
Definicja
Leukodyst
rofie (LD) to nazwa stosowana na określenie wszystkich genetycznie uwarunkowanych chorób z przeważającym zajęciem istoty białej. 1 Niektórzy używają tego pojęcia w odniesieniu do chorób istoty białej, w których zaburzenia mielinizacji są wynikiem patologii komórek glejowych, ponadto wyróżniają genetycznie uwarunkowane leukoencefalopatie (gLE), w których istota biała jest uszkodzona wtórnie do zajęcia innych elementów tkanki mózgowej (np. naczyń) lub zaburzeń ogólnoustrojowych (najczęściej metabolicznych) albo dotyczących układu nerwowego. Liczbę tych chorób ocenia się łącznie na ponad 100. 2, 3, 4 Zarówno w LD, jak i gLE uszkodzeniu ośrodkowego układu nerwowego (OUN) towarzyszy niekiedy neuropatia obwodowa. Według międzynarodowych ustaleń ścisłe kryteria włączenia do LD spełnia ponad 30 jednostek chorobowych, a do gLE – ponad 60. 2, 3, 4 Obecnie w ok. 80% przypadków udaje się ustalić podłoże molekularne. 1, 2
Rys historyczny
Pojęcie leukodystrofii wprowadził Max Bielschowsky w 1928 r., a utrwalił Franz Seitelberger w 1984 r. na określenie genetycznie uwarunkowanych chorób z zajęciem mieliny. 5 Początkowo do ich rozpoznania konieczne były badania neuropatologiczne, które niestety tylko w nielicznych przypadkach wyjaśniały podłoże choroby (np. komórki globoidalne typowe dla choroby Krabbego). Ze względu na nieswoisty obraz mikroskopowy większość LD była klasyfikowana – zależnie od metod barwienia – jako ortochromatyczna, metachromatyczna czy sudanofilna. 5 Z czasem kilka najczęstszych, np. leukodystrofię metachromatyczną (MLD – metachromatic leukodystrophy), chorobę Krabbego czy adrenoleukodystrofię, zaczęto rozpoznawać przyżyciowo dzięki badaniom biochemicznym. 2, 5 Wraz z wprowadzeniem metod neuroobrazowania metodą rezonansu magnetycznego (MR – magnetic resonance) i rozwojem badań molekularnych dokonał się szybki postęp w rozpoznawaniu tych chorób. Poznano zarówno podłoże molekularne dobrze już znanych jednostek, np. choroby Alexandra (ALXDRD – Alexander disease), jak i wiele nowych LD. 2, 6, 7
W ostatniej dekadzie metody sekwencjonowania nowej generacji zrewolucjonizowały diagnostykę i obecnie w przypadku coraz większej liczby tych chorób można ustalić przyczynę. 2, 3 Wiedza o patologii na poziomie subkomórkowym poszerzyła się na tyle, że u podłoża niektórych LD i gLE oprócz defektów białkowych wykryto inne zaburzenia. Są to m.in. zaburzenia odnowy DNA czy defekty cząsteczek RNA biorących udział w metabolizmie podstawowym komórek, w regulacji budowy, funkcji oraz utrzymaniu prawidłowego składu osłonki mielinowej. 2, 4
Epidemiologia i zagadnienia genetyczne
LD i gLE to choroby rzadkie, a nawet bardzo rzadkie. Częstość występowania jest różna dla poszczególnych jednostek chorobowych. 7 W każdym przypadku podejrzenia LD i gLE zaleca się trzypokoleniową analizę rodowodu, która może ukierunkować rozpoznanie poprzez naprowadzenie na tryb dziedziczenia. LD i gLD zazwyczaj są dziedziczone w sposób mendlowski – autosomalnie recesywnie, rzadziej autosomalnie dominująco lub recesywnie w sprzężeniu z chromosomem X. Choroby dziedziczone recesywnie występują w jednym pokoleniu u obu płci, dziedziczone dominująco – w kilku pokoleniach, również niezależnie od płci, ale mogą pojawić się pozornie sporadycznie wskutek nowej mutacji, tak jak większość przypadków ALXDRD.
W przypadku LD sprzężonych z chromosomem X chorują najczęściej chłopcy, jednak także matki nosicielki mogą mieć lżejsze objawy choroby. W chorobach dziedziczonych po linii matczynej, wywołanych mutacjami mitochondrialnego DNA (mtDNA), takich jak zespół Kearnsa-Sayre’a czy zespół MELAS (mitochondrial myopathy, encephalopathy, lactic acidosis, stroke-like episodes), na który składają się miopatia mitochondrialna, encefalopatia, kwasica mleczanowa i występowanie incydentów podobnych do udarów, może być również zajęta istota biała, jednak nie są one zaliczane do LD. W przypadku dziedziczenia odmatczynego matka nosicielka może przekazać zmutowane cząsteczki mtDNA całemu potomstwu, jednak w różnym odsetku. Ryzyko zachorowania dotyczy dzieci obu płci, u których odsetek cząsteczek mtDNA z mutacją przekracza 65-90%; chorobę mogą przekazać wyłącznie jej córki.
Ponieważ większość LD uwarunkowana jest autosomalnie recesywnie, wczesne ustalenie rozpoznania przyczynowego może zapobiec urodzeniu się kolejnych chorych dzieci w rodzinie.
Klasyfikacja leukodystrofii. Rola badań neuroobrazowych
Na podstawie badań metodą MR wyróżnia się dwie grupy LD: hipomielinizacyjne (HLD – hypomyelinating leukodystrophy) i demielinizacyjne (LDD – leukodystrophy demyelinating). W pierwszej grupie brakuje mielinizacji, jest ona trwale upośledzona i/lub nieprawidłowa (dysmielinizacja), natomiast w drugiej grupie dochodzi do postępującego uszkodzenia mieliny. 3, 6 Ostatnio jednak van der Knaap zaproponowała, aby rozszerzyć klasyfikację i oprzeć ją na kryteriach neuropatologicznych, w zależności od tego, które elementy istoty białej są pierwotnie i w dominujący sposób uszkodzone. W tym podziale wyróżnia się LD z uszkodzeniem mieliny (hipomielinizacyjne, demielinizacyjne, z wakuolizacją mieliny), leukoaksonopatie, astrocytopatie, mikroglejopatie oraz leukowaskulopatie. 1
Przyżyciową ocenę rodzaju zaburzeń umożliwia badanie neuroobrazowe metodą MR wykonane w sekwencjach T2 (w tym T2-FLAIR), T1, SWI oraz DWI. W ostatnich dekadach opracowano wzorce charakterystyczne dla wielu poszczególnych LD i gLE, ułatwiające diagnostykę różnicową. 8, 9, 10 Zmianom hipomielinizacyjnym w MR odpowiada lekko hiperintensywny sygnał istoty białej w stosunku do kory w sekwencjach T2 (zwłaszcza w T2-FLAIR) oraz izo-, hipo- lub hiperintensywny w T1. Dysmielinizację cechują słabe zróżnicowanie korowo-podkorowe oraz nieostro odgraniczone zmiany w istocie białej o różnej intensywności sygnału (patchy – pstre). Wreszcie demielinizacji odpowiada silnie hiperintensywny sygnał istoty białej w T2, natomiast hipointensywny w T1. 8, 9, 10
Klasyfikacja ta, jak każda inna, jest uproszczona i zależnie od wielu czynników, w tym od zaawansowania choroby i wieku chorego, różne zmiany mogą się na siebie nakładać. W postaciach niemowlęcych i wczesnodziecięcych LD demielinizacja nakłada się na hipomielinizację, ponieważ w tym wieku mielinizacja jest nieukończona. Niekiedy prowadzi to do błędnej interpretacji obrazu. Za kryterium rzeczywistej hipomielinizacji przyjęto utrzymywanie się zmian w kolejnych badaniach MR wykonywanych >12 m.ż. w okresie przynajmniej 6 miesięcy. 8, 9 Nie powinno się traktować jako LD opóźnienia mielinizacji, będącej nieswoistym objawem w wielu przypadkach opóźnienia psychoruchowego, jeśli w kolejnych badaniach obserwuje się jej poprawę. W piśmiennictwie medycznym dostępne są wzorce mielinizacji zależne od wieku. 8, 9
Zmiany w LD i gLE są na ogół symetryczne. W diagnostyce różnicowej duże znaczenie ma staranna analiza rozległości i lokalizacji zmian w MR. Istotne jest stwierdzenie, czy została zajęta cała istota biała, czy też np. głównie okołokomorowa, głęboka lub podkorowa; czy przeważa zajęcie przednich okolic mózgu, czy tylnych; czy po podaniu środka kontrastowego uwidacznia się wzmocnienie sygnału na obwodzie zmian. 8, 9 Istotne w różnicowaniu mogą być również współistniejące uszkodzenia innych struktur mózgowia i rdzenia. 9, 10 Sygnałem obecności zwapnień lub odkładania żelaza jest hiperintensywny sygnał w sekwencji T1 oraz hipointensywny w SWI, nie ma więc potrzeby wykonywania tomografii komputerowej w celu ich wykrycia. Spektroskopia rezonansu magnetycznego (MRS – magnetic resonance spectroscopy) może ukazać charakterystyczne dla danej jednostki chorobowej metabolity, jak np. „pik” N-acetyloasparaginianów (NAA) w chorobie Canavan (CD – Canavan disease), mleczanów w chorobach mitochondrialnych czy lipidów w zespole Sjögrena-Larssona. Hiperintensywność w sekwencji DWI to cecha typowa dla aktywnego procesu chorobowego. 8, 9 Na ogół jednak do ustalenia rozpoznania niezbędne jest skorelowanie zmian neuroobrazowych z obrazem klinicznym.
Budowa istoty białej
W istocie białej OUN są przede wszystkim liczne drogi nerwowe zbudowane z aksonów. Z elementów komórkowych występują w niej komórki glejowe – oligodendrocyty, astrocyty włókniste oraz mikroglej. Znajdują się tu również bogata sieć naczyń krwionośnych oraz substancja międzykomórkowa. 5
Oligodendrocyty wytwarzają osłonki mielinowe oraz odgrywają rolę w budowie, utrzymywaniu prawidłowej ilości i składu mieliny oraz w procesach jej odnawiania. Osłonki mielinowe, które są zmodyfikowanymi wypustkami oligodendrocytów, zapewniają izolację konieczną do prawidłowego działania dróg nerwowych. W skład osłonki mielinowej wchodzą głównie tłuszcze złożone – fosfolipidy (40-50%), galaktolipidy (27-30%) oraz cholesterol (25%). Szczególną cechą istoty białej, w odróżnieniu od szarej, jest obecność cerebrozydów, należących do galaktolipidów. Spośród białek mieliny dwa główne to lipoproteina 1 (PLP1 – proteolipid protein 1), stanowiąca ok. 50%, oraz podstawowe białko mieliny (MBP – myelin basic protein), stanowiące ok. 30-35%. Pozostałe to m.in. liczne glikoproteiny, inne lipoproteiny oraz inne białka oligodendrocytów. 5
Astrocyty są najliczniejszymi komórkami glejowymi mózgu i pełnią wiele ważnych funkcji, w tym strukturalne, odżywcze, naprawcze, a także regulujące przepływ krwi (przez tzw. falę Ca++) i modulujące neuroprzekaźnictwo. Biorą udział w tworzeniu bariery krew–mózg. Otaczają synapsy, uczestnicząc w przemianie neurotransmiterów, zwłaszcza glutaminianów i kwasu γ-aminomasłowego (GABA – γ-aminobutyric acid). Pośredniczą także w wymianie różnych składników między naczyniami krwionośnymi a tkanką mózgową, wydzielają kilka czynników wzrostowych oraz mają znaczenie w utrzymaniu równowagi wodno-elektrolitowej i stałości środowiska tkanki mózgowej. Ich wypustki otaczają przewężenia Ranviera, zapewniając izolację konieczną do szybkości przewodzenia bodźca elektrycznego. Ostatnio pojawiły się doniesienia, że odgrywają też rolę w realizacji funkcji poznawczych. W warunkach fizjologicznych w istocie białej przeważają astrocyty włókniste. 5
Komórki mikrogleju to specyficzne dla OUN komórki pochodzenia mezenchymalnego, biorące udział w odpowiedzi immunologicznej, kontrolujące homeostazę oraz będące źródłem makrofagów mózgowych i cytokin.
Patogeneza leukodystrofii i genetycznie uwarunkowanych leukoencefalopatii
Patogeneza LD i gLE jest bardzo złożona i różna dla każdej z jednostek chorobowych i podgrup chorób. Jak wspomniano wyżej, u podłoża LD leżą zarówno niedobory białek biorących udział w budowie, odnawianiu oraz funkcjonowaniu mieliny, jak i defekty cząsteczek RNA uczestniczących w metabolizmie podstawowym komórek. Patologia może również wynikać z uszkodzenia przez różne spichrzane metabolity lub endogenne substancje toksyczne (np. psychozyna w chorobie Krabbego). W wielu współistnieje czynnik zapalny, np. udział cytokin pochodzących z mikrogleju w patogenezie adrenoleukodystrofii sprzężonej z chromosomem X (X-ALD – X-linked adrenoleukodystrophy), a także w chorobie Krabbego. 1
W gLE uszkodzenie może wynikać z: 1. patologii naczyń (np. mózgowa dominująca arteriopatia z udarami podkorowymi, leukoencefalopatią i otępieniem [CADASIL – cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy], arteriopatia mózgowa dziedziczona autosomalnie recesywnie z zawałami podkorowymi i encefalopatią [CARASIL – cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy], choroba Fabry’ego); 2. toksycznego działania produktów przemian pośrednich (np. aminoacydopatie, kwasice organiczne); 3. zmian w istocie pozakomórkowej (np. choroba ze spichrzaniem kwasu sjalowego i niektóre rzadkie kolagenopatie); 4. zaburzonej odnowy DNA (np. zespół 4H), 5. zaburzeń energetycznych (choroby mitochondrialne) i innych. 1, 3, 4
Diagnostyka leukodystrofii
W obrazie klinicznym LD i gLE zwraca uwagę przewaga zaburzeń ruchowych w początkowym okresie choroby. Z reguły jest tak, że im wcześniej wystąpi choroba, tym cięższy i bardziej nieswoisty ma przebieg.
W chorobach z hipomielinizacją (HLD) przebieg kliniczny jest powolniejszy. Rozwój psychoruchowy, zwłaszcza ruchowy, zwykle jest opóźniony, a u chorych najczęściej stwierdza się uogólnioną wiotkość przy zachowanych odruchach. Prawie zawsze występuje oczopląs. Niekiedy obserwuje się okresy pozornej poprawy i długie fazy plateau, co może się przyczynić do błędnego rozpoznania mózgowego porażenia dziecięcego. Rozwój emocjonalny, społeczny i intelektualny jest względnie lepszy, a niekiedy prawidłowy. 11, 12 Z kolei w leukodystrofiach demielinizacyjnych (LDD) objawy występują z reguły po okresie prawidłowego rozwoju i mają szybszy przebieg. Zależnie od wieku obserwuje się w nich regres rozwoju/otępienie, ataksję i spastyczność. Współistnienie neuropatii obwodowej może powodować przejściową wiotkość.
W różnicowaniu bywa pomocna analiza okoliczności wystąpienia choroby: początek prawie nieuchwytny (jak np. w chorobach lizosomalnych) czy ostry, np. po szczepieniu, infekcji (jak w chorobach mitochondrialnych), urazie głowy czy gorączce (jak w leukodystrofii ze znikającą istotą białą [VWM – vanishing white matter]), a także analiza jej przebiegu (powoli postępujący czy falujący). 11, 12, 13 Na właściwe rozpoznanie może również naprowadzić obecność objawów towarzyszących ze strony innych narządów i układów. Szczególnie wartościowe jest stwierdzenie oprócz cech hipoplazji lub zaniku nerwów wzrokowych dodatkowych zmian w narządzie wzroku (zaćma, retinopatia, zmętnienie rogówki).
Do ustalenia rozpoznania przyczynowego niezbędne są liczne badania laboratoryjne, w tym biochemiczne (np. ocena aktywności resztkowej enzymu, wykrycie nieprawidłowych metabolitów w płynach ustrojowych), elektrofizjologiczne, rzadziej – histo- i immunohistochemiczne hodowanych fibroblastów lub tkanek pobranych drogą biopsji. 11, 12, 13 Biopsja, będąca badaniem inwazyjnym, zalecana jest coraz rzadziej, poza tym wymaga oceny w wyspecjalizowanych laboratoriach. Jak wspomniano wyżej, badania MR są podstawą w diagnostyce LD i gLE, jednak zawsze konieczna jest staranna analiza kliniczno-neuroobrazowa. 11, 12, 13
Obecnie coraz częściej zaleca się badania molekularne w celu znalezienia patogennej mutacji w DNA. Koszty tych badań maleją, więc powinno się rozważyć ich wykonanie nawet na początku procedur diagnostycznych. Znalezienie mutacji nie tylko ostatecznie potwierdza przyczynę choroby, ale też umożliwia objęcie pacjenta i jego rodziny poradnictwem genetycznym. Pozwala również wdrożyć właściwe dla danej jednostki chorobowej postępowanie wspierające, rehabilitacyjne i zapobiegające powikłaniom, a co za tym idzie – zmniejszyć publiczne nakłady na lecznictwo. 14, 15
Leukodystrofie hipomielinizacyjne (HLD)
HLD są spowodowane trwałym brakiem mieliny, jej niedoborem i/lub nieprawidłową mielinizacją. Występują głównie u dzieci i młodzieży. Zależnie od patogenności mutacji mogą się ujawnić począwszy od okresu płodowego. Do dziś poznano ponad 14 genów leżących u podłoża HLD, ale wśród tego typu zaburzeń wciąż dominują choroby o nieustalonej etiologii. 3, 6 Leukodystrofie z dysmielinizacją (dysLD), czyli z niedostateczną i/lub nieprawidłową mielinizacją, wynikają najczęściej z aberracji chromosomowych.
Dysmielinizacja w obrazie MR stwierdzana jest również w łagodniejszych postaciach chorób zaliczanych do HLD, jak postać alleliczna choroby Pelizaeusa-Merzbachera (PMD – Pelizaeus-Merzbacher disease) – parapareza spastyczna typu 2 (SPG2 – spastic paraplegia type 2), a także w niektórych innych paraparezach spastycznych (m.in. SPG35).
W postaciach wrodzonych, które zaczynają się w okresie płodowym, obserwuje się najcięższy i niepomyślny przebieg. Chore dzieci są znacznie opóźnione psychoruchowo, na ogół bardzo wiotkie i – o ile nie rozwinęła się neuropatia obwodowa – mają zachowane odruchy. Niemal zawsze występuje oczopląs. W niektórych HLD pojawia się również padaczka. Szybko dochodzi do uspastycznienia i śmierci wskutek powikłań.
Postacie o późniejszym początku charakteryzują się nieswoistym, niekiedy nieomal stacjonarnym lub bardzo wolno postępującym przebiegiem, z przeważającym opóźnieniem ruchowym na skutek wiotkości. Wiotkość wynika głównie z uszkodzenia OUN, ale może być też efektem współistniejącej neuropatii obwodowej. Do zespołu piramidowego, a zwłaszcza spastyczności wynikającej z uszkodzenia istoty białej, dochodzi po wielu miesiącach, a nawet latach. Oczopląs jest nieomal stałym objawem, choć jego nasilenie może się zmniejszać z wiekiem. Inaczej niż w przypadku LDD prawie zawsze współistnieją objawy pozapiramidowe. W początkowym okresie przeważają dyskinezy, w miarę postępowania choroby pojawia się dystonia. Odruchy głębokie są zachowane, a dopiero z czasem i nie we wszystkich przypadkach stają się one wygórowane. W razie neuropatii mogą być osłabione. Objaw Babińskiego pojawia się nie zawsze i zazwyczaj po dłuższym okresie choroby. Rozwój umysłowy, emocjonalny i społeczny dzieci z HLD i dysLD jest względnie lepszy niż ruchowy, a niekiedy nawet prawidłowy. Stężenie białka w płynie mózgowo-rdzeniowym nie jest podwyższone, czasami bywa wręcz niskie.
Badanie MR ujawnia zmiany charakterystyczne dla hipomielinizacji/dysmielinizacji (najczęściej niewielka lub nierównomierna hiperintensywność istoty białej w sekwencji T2, zwłaszcza w T2-FLAIR, w stosunku do kory mózgu i zwojów podstawy). 9, 10 W niektórych HLD współistnieją zmiany w zwojach podstawy oraz zanik móżdżku, jak w HLD typu 6 uwarunkowanej mutacją genu TUBB4A. Ponadto na prawidłowe rozpoznanie może naprowadzić współistnienie zmian w innych narządach i układach, takich jak np. zaćma w HLD typu 4 uwarunkowana mutacją HSPD1 czy hipodoncja i/lub hipogonadyzm hipogonadotropowy w HLD typu 7 i 8. 11, 12, 13
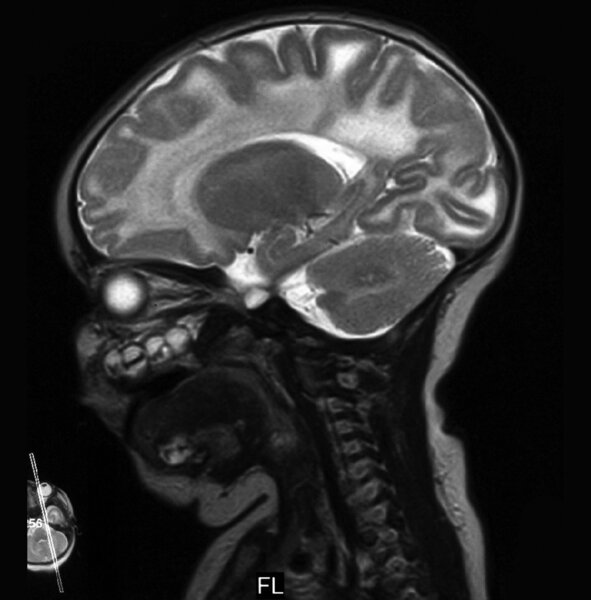
Choroba Pelizaeusa-Merzbachera (PMD)
PMD to najczęstsza i najlepiej poznana leukodystrofia hipomielinizacyjna (HLD1). Częstość jej występowania ocenia się na 1-2/100 000 urodzeń. Spowodowana jest mutacją genu PLP1 i dziedziczona recesywnie w sprzężeniu z chromosomem X. Wspomniany gen koduje białko PLP1 oraz izoformę DM20. PMD dotyka najczęściej chłopców, u których może mieć postać wrodzoną, klasyczną lub SPG2. Ta ostatnia może występować u kobiet nosicielek mutacji. W badaniu MR mózgowia stwierdza się rozległą hipomielinizację istoty białej mózgu z zajęciem torebek wewnętrznych, pnia mózgu i móżdżku (ryc. 1).
We wrodzonej postaci PMD, niezwykle rzadkiej, obserwuje się hipotrofię wewnątrzmaciczną, znaczne opóźnienie rozwoju psychoruchowego i fizycznego, skrajną wiotkość, w tym stridor, wrodzone małogłowie oraz oczopląs; odruchy głębokie są zachowane. Szybko dochodzi do niewydolności oddechowej i zgonu.
W klasycznej postaci PMD od okresu niemowlęcego obserwuje się oczopląs, uogólnioną wiotkość oraz opóźnienie rozwoju, zwłaszcza ruchowego. Ponadto występują ataksja móżdżkowa oraz dyskinezy, z czasem dołącza się dystonia. Odruchy głębokie są często wygórowane. Ze względu na powolny przebieg i poprawę niektórych parametrów rozwojowych u wielu chorych z klasyczną postacią PMD może być błędnie rozpoznawana postać móżdżkowa lub pozapiramidowa mózgowego porażenia dziecięcego. W tej postaci dopiero po kilku latach dochodzi do spastyczności. Rozwój umysłowy jest różny: od upośledzenia głębokiego do prawidłowego. W badaniu okulistycznym stwierdza się blade tarcze nerwów wzrokowych, jednak widzenie jest zachowane względnie długo. Najczęstszy typ patogennej mutacji stanowi duplikacja całego genu PLP1. W badaniach z ostatnich lat nie potwierdzono korelacji fenotyp-genotyp – mutacje punktowe i duplikacje całego genu PLP1 wykryto u podłoża ciężkich wrodzonych postaci PMD, ale też względnie łagodnych klasycznych, w tym u chorych z prawidłowym rozwojem umysłowym. 7
Leukodystrofia z hipomielinizacją oraz zanikiem zwojów podstawy i móżdżku (HLD6, H-ABC)
Leukodystrofia z hipomielinizacją oraz zanikiem zwojów podstawy i móżdżku (HLD6; H-ABC – hypomyelination with atrophy of basal ganglia and cerebellum) to niezwykle rzadka, sporadycznie występująca choroba wywołana dominującymi mutacjami w jednym allelu genu TUBB4.
W postaci wrodzonej ruchy płodów relacjonowane są jako osłabione; może również występować wielowodzie. Dzieci rodzą się z prawidłową masą i obwodem głowy, jednak przyrost obwodu głowy jest spowolniony i rozwija się małogłowie. Oczopląs, wiotkość oraz opóźnienie psychoruchowe obserwuje się od pierwszych miesięcy życia. Z czasem pojawia się narastający zespół piramidowo-pozapiramidowy ze spastycznością, dystonią, rzadziej z choreoatetozą. Odruchy głębokie są wygórowane. Niekiedy może wystąpić padaczka. W postaciach o późniejszym przebiegu często rozpoznawana jest błędnie postać piramidowo-pozapiramidowa mózgowego porażenia dziecięcego. Rozwój umysłowy jest najczęściej nieprawidłowy.
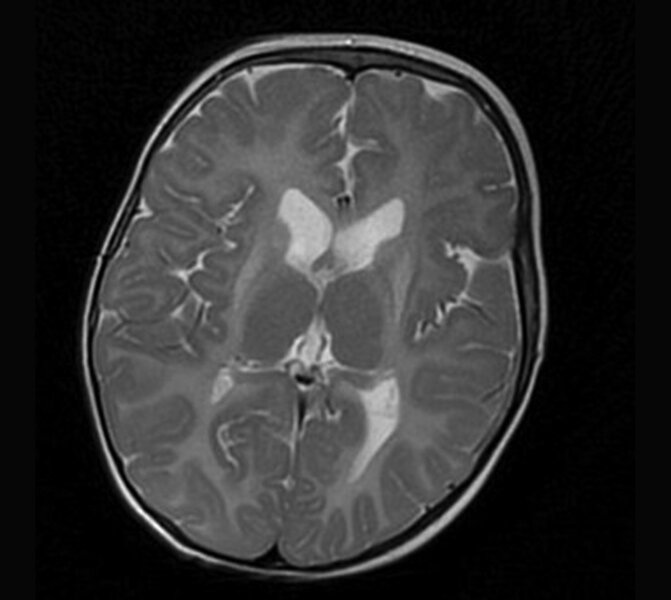
Rycina 2. Leukodystrofia hipomielinizacyjna z zanikiem zwojów podstawy i móżdżku (HLD6). MR, sekwencja T2. Rozległa hipomielinizacja oraz hiperintensywny sygnał zanikających, obkurczonych skorup. Dziewczynka 14-miesięczna z zespołem piramidowo-pozapiramidowym
Na rozpoznanie pozwala badanie MR, w którym oprócz rozległej hipomielinizacji stwierdza się postępujący zanik prążkowia (skorup i głów jąder ogoniastych), a z czasem również móżdżku (ryc. 2). Opisano postacie HLD6 wyłącznie z LD, bez zaniku zwojów podstawy. Choroba ma charakter powoli postępujący i zależnie od ciężkości przebiegu w 2-3 dekadzie życia dochodzi do zgonu. 3, 4, 6
Leukodystrofie hipomielinizacyjne typu 7 i 8
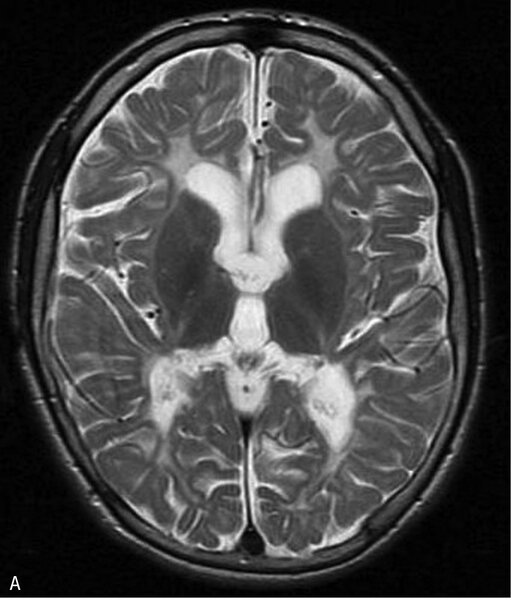
Rycina 3. Leukodystrofia hipomielinizacyjna typu 7 (HLD7) z hipodoncją i hipogonadyzmem hipogonadotropowym. A. MR, sekwencja T2. Hipomielinizacja oraz zmniejszona objętość całej istoty białej półkul mózgu. B. Ścieńczałe ciało modzelowate, widoczny również zanik móżdżku. Chłopiec 10-letni z powoli postępującym zespołem pozapiramidowo-piramidowym i ataksją
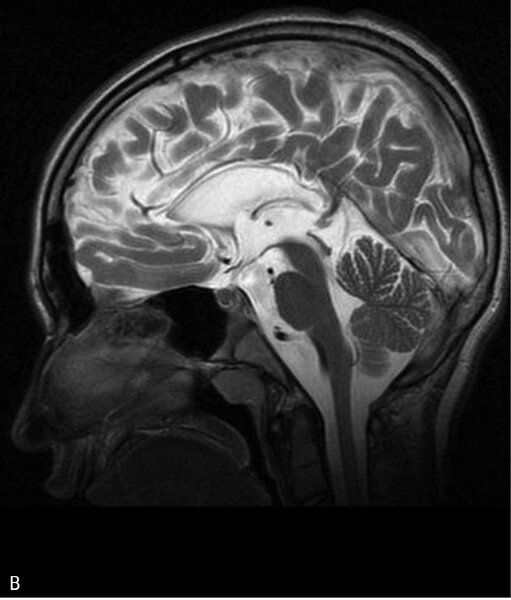
Rycina 3. Leukodystrofia hipomielinizacyjna typu 7 (HLD7) z hipodoncją i hipogonadyzmem hipogonadotropowym. A. MR, sekwencja T2. Hipomielinizacja oraz zmniejszona objętość całej istoty białej półkul mózgu. B. Ścieńczałe ciało modzelowate, widoczny również zanik móżdżku. Chłopiec 10-letni z powoli postępującym zespołem pozapiramidowo-piramidowym i ataksją
Leukodystrofie hipomielinizacyjne typu 7 i 8 (HLD7, HLD8) są związane z mutacjami w genach POLR3A i POLR3B, kodujących polimerazy RNA. Pełną postać określa się mianem zespołu 4H, ponieważ oprócz rozległej hipomielinizacji mózgowia stwierdza się hipodoncję (brak zawiązków niektórych zębów) oraz hipogonadyzm hipogonadotropowy. U wszystkich chorych po kilku latach dochodzi do zmniejszenia objętości ciała modzelowatego oraz zaniku móżdżku. Opisano postacie poronne HLD7/8 wykryte dzięki badaniom genetycznym, bez rozległej hipomielinizacji, jednak u tych chorych w badaniu MR stwierdzano najczęściej zajęcie odnóg tylnych torebek wewnętrznych i/lub zanik móżdżku (ryc. 3). Hipodoncja i zaburzenia hormonalne występowały rzadziej. HLD7/8 mają najczęściej powolny, postępujący przebieg, jednak z reguły im wcześniejsze ujawnienie choroby, tym ten przebieg jest cięższy.
Klinicznie w zespole 4H obserwuje się różnego stopnia opóźnienie rozwoju ruchowego, niezgrabność ruchową, uogólnioną wiotkość oraz powoli postępującą ataksję. Rozwój umysłowy jest opóźniony w różnym stopniu. Odruchy głębokie są osłabione; dopiero po latach pojawia się objaw Babińskiego. Ząbkowanie jest najczęściej opóźnione, brakuje też niektórych zawiązków zębów mlecznych i stałych. Sporadycznie opisywano ząbkowanie noworodkowe. U chorych z zespołem 4H nie pojawiają się samoistnie cechy dojrzewania płciowego i towarzyszy temu niedobór wzrostu, co może prowadzić do błędnego podejrzenia wielohormonalnej niedoczynności przysadki lub zespołu Turnera u dziewcząt. Zależnie od ciężkości przebiegu choroby narastają zaburzenia neurologiczne i regres umysłowy lub otępienie. W postaciach poronnych choroba może się ujawnić nawet w 2 dekadzie życia w postaci narastających zaburzeń chodu i ataksji, a na rozpoznanie może naprowadzić hipodoncja. 16 Leczenie jest objawowe. W postaciach pełnoobjawowych wskazana jest suplementacja hormonalna.
Delecja 18q(-)
Delecja 18q(-) to dysLD związana z brakiem MBP, dla którego gen znajduje się w terminalnym odcinku długiego ramienia chromosomu 18. Obraz kliniczny jest różny w zależności od wielkości delecji i związanego z tym ubytku materiału genetycznego. Obserwuje się upośledzenie psychoruchowe lub niezgrabność ruchową, małogłowie, różne cechy dysmorficzne twarzoczaszki, w tym uszu (łącznie z zarośnięciem przewodu słuchowego i niedosłuchem lub głuchotą), niekiedy wady serca, rzadziej wady kończyn i narządów płciowych. W cięższych postaciach występują oczopląs, ataksja i zespół pozapiramidowo-piramidowy. Stężenie białka w płynie mózgowo-rdzeniowym jest prawidłowe.
Obraz MR mózgu charakteryzuje się słabym zróżnicowaniem sygnału kory i istoty białej z nieostro odgraniczonymi ogniskami o różnej intensywności sygnału, niekiedy jakby rozmytymi w sekwencji T2, FLAIR oraz T1; może występować zanik móżdżku. W diagnostyce niezbędne są badania genetyczne. 3, 7
Wrodzone choroby istoty białej z demielinizacją
LDD to choroby spowodowane postępującym uszkodzeniem mieliny. Zazwyczaj ich przyczynę stanowią wrodzone błędy metaboliczne. 1, 3, 12, 13, 15 W Polsce i na świecie najczęstsze są choroby lizosomalne – leukodystrofia metachromatyczna (MLD) i leukodystrofia globoidalna [GLD – globoid cell leukodystrophy]), peroksyzomalne (X-ALD) oraz mózgowe kwasice organiczne (CD). Do tej grupy zaliczają się też postacie ALXDRD i VWM o późnym początku, jak również gLE z uszkodzeniem mieliny wtórnym do innych zaburzeń, niezależnych od dysfunkcji komórek glejowych.
Objawy kliniczne poszczególnych LDD nie są swoiste, a przebieg jest zwykle tym cięższy, im wcześniej te objawy się ujawnią. Po okresie prawidłowego rozwoju u niemowląt i małych dzieci dochodzi do regresu psychoruchowego, u starszych – do zaburzeń chodu i ataksji. Jeśli współistnieje neuropatia obwodowa, obserwuje się przejściową wiotkość. Z czasem, w ciągu kilku lub kilkunastu miesięcy, pojawiają się spastyczny niedowład czterokończynowy, zaburzenia połykania i odmóżdżenie. U dorosłych dochodzi do ataksji, otępienia i spastyczności, a objawy kliniczne postępują w ciągu kolejnych lat.
W płynie mózgowo-rdzeniowym w okresie aktywnego rozpadu mieliny stwierdza się podwyższone stężenie białka. Diagnostyka opiera się na analizie obrazu klinicznego, badaniach neuroobrazowych, które również wymagają precyzyjnej analizy umiejscowienia zmian i ich charakteru, badaniach biochemicznych, elektrofizjologicznych, a ostatnio także na badaniach genetycznych. 11, 12
Leukodystrofia metachromatyczna (MLD)
MLD to najczęstsza LDD w Polsce i na świecie – częstość jej występowania ocenia się na 1/20 000 urodzeń. Uwarunkowana jest autosomalnie recesywnie mutacjami genu ARSA i należy do chorób lizosomalnych. Obserwuje się pełne spektrum postaci, od niemowlęcych do obserwowanych u dorosłych. Przebieg u niemowląt i małych dzieci jest najczęściej szybki i niepomyślny. Im wcześniej ujawni się choroba, tym cięższy przebieg. Po etapie zatrzymania rozwoju w ciągu kilku tygodni dochodzi do regresu, spastyczności i odmóżdżenia. U dzieci starszych po okresie zaburzeń chodu, ataksji, niekiedy przejściowej wiotkości wskutek neuropatii obwodowej w ciągu kilku miesięcy rozwija się spastyczny niedowład czterokończynowy. U młodzieży i dorosłych choroba przebiega wolniej, a objawom piramidowym towarzyszą ataksja i postępujące otępienie. Występują również postacie z izolowanym otępieniem.
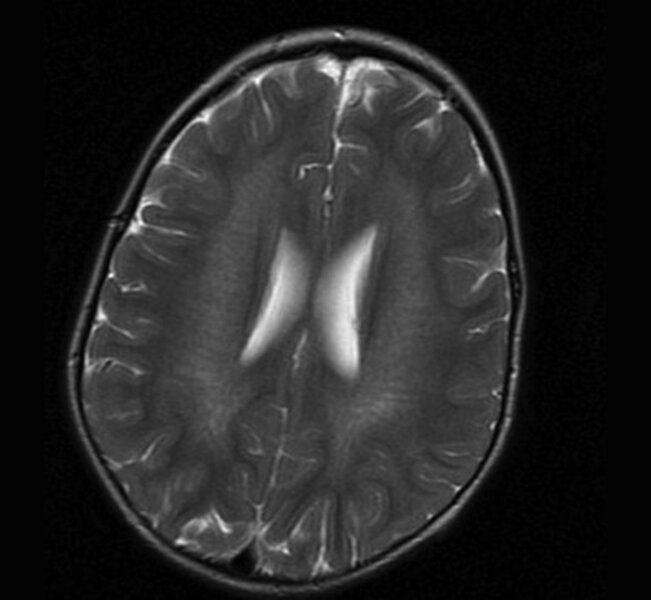
Rycina 4. Leukodystrofia metachromatyczna (MLD) potwierdzona molekularnie. MR, sekwencja T2. W istocie białej głębokiej widoczne jeszcze zachowane włókna nerwowe u 3-letniego chłopca z 3-miesięcznym wywiadem chorobowym i objawami ataksji móżdżkowej
Badania neuroobrazowe ukazują obustronne zmiany demielinizacyjne głębokiej istoty białej. W początkowej fazie choroby widoczne są zachowane włókna nerwowe (ryc. 4). Diagnostyka opiera się na stwierdzeniu znacznie obniżonej aktywności enzymu arylosulfatazy A, a ostatnio coraz częściej na badaniach genetycznych. Obserwuje się korelację genotypowo-fenotypową. 3, 7
Leukodystrofia globoidalna Krabbego (GLD)
GLD to również dziedziczona autosomalnie recesywnie choroba lizosomalna wynikająca z mutacji genu GACL. W Polsce jest rzadsza niż MLD i występuje z częstością 1/40 000. Może się ujawnić począwszy od okresu niemowlęcego do wieku dorosłego. Przebieg GLD jest tym cięższy, im wcześniej choroba da objawy.
U niemowląt obserwuje się szybki regres psychoruchowy oraz trudny do opanowania niepokój – dzieci sprawiają wrażenie cierpiących, przestają jeść, szybko rozwijają się tetrapareza spastyczna i odmóżdżenie. U dzieci starszych przebieg jest nieco wolniejszy. Po przejściowym okresie zatrzymania rozwoju, ataksji oraz wiotkości wskutek demielinizacyjnej neuropatii obwodowej rozwija się postępujący niedowład spastyczny czterokończynowy, następuje regres rozwoju i w ciągu paru lat dochodzi do zgonu. Postacie typu dorosłe cechują się ataksją, spastycznością z przewagą kończyn dolnych i otępieniem.
W obrazie MR u niemowląt stwierdza się rozlane zmiany w istocie białej zaczynające się w pniu mózgu i móżdżku, tj. w strukturach już zmielinizowanych, a po kilku miesiącach w obu półkulach; sygnał wzgórz jest nieco hiperintensywny; nerwy wzrokowe są pogrubiałe. U starszych dzieci widoczne są rozległe zmiany w istocie białej półkul z zaoszczędzeniem U-włókien, a w postaciach dorosłych – przewaga zajęcia płatów ciemieniowo-potylicznych. Nie obserwuje się korelacji fenotyp-genotyp, w jednej rodzinie mogą występować różne postacie choroby.
Uważa się, że w GLD patogenną rolę odgrywa psychozyna (metabolit cerebrozydów), która uszkadza oligodendrocyty, co prowadzi do demielinizacji. Ostatnio zwrócono też uwagę na inicjującą rozpad mieliny rolę prozapalnych cytokin, wydzielanych przez mikroglej. Komórki globoidalne charakterystyczne dla GLD widoczne są w badaniu mikroskopowym w obszarach rozpadu mieliny. W diagnostyce stosuje się badania biochemiczne (ocena aktywności resztkowej galaktocerebrozydazy) i coraz częściej badania genetyczne. 3, 7, 12
Należy pamiętać, że istnieją rzadkie postacie MLD i GLD, których przebieg jest podobny jak w chorobach klasycznych, uwarunkowane deficytami kofaktorów – sapozyn. Wymagane są celowane badania molekularne w razie silnego podejrzenia tych postaci. 7, 13
Adrenoleukodystrofia sprzężona z chromosomem X (X-ALD)
X-ALD to druga co do częstości LDD w Polsce. Częstość jej występowania ocenia się na 1/20 000-50 000 urodzeń. X-ALD to choroba peroksyzomalna, uwarunkowana recesywną mutacją genu ABCD1, który koduje białko błony peroksyzomalnej. Jest to transporter przenoszący kwasy tłuszczowe o bardzo długim łańcuchu (VLCFA – very long chain fatty acids) do wnętrza organelli.
Wyróżnia się postaci mózgowe X-ALD, występujące głównie u chłopców w wieku 4-12 lat, rzadziej u młodych mężczyzn, oraz adrenomieloneuropatię, notowaną głównie u dorosłych. Każdej z tych postaci może towarzyszyć niewydolność kory nadnerczy (>60% chorych), która bywa niekiedy jedynym, izolowanym objawem. U dorosłych chorych możliwe są impotencja oraz niewydolność gonad. U kobiet nosicielek mutacji objawy X-ALD mogą być łagodniejsze. Postać mózgowa manifestuje się zaburzeniami chodu, ataksją, niekiedy drgawkami, a następnie szybko postępującym (kilka miesięcy) zespołem porażenia czterokończynowego z wygórowanymi odruchami i spastycznością. Objawom tym towarzyszą często zaburzenia emocjonalne oraz zaburzenia zachowania, rzadziej objawy psychotyczne. Zwykle po kilku miesiącach dochodzi do zespołu odmóżdżenia; średnie przeżycie chorych z postacią mózgową wynosi 2 lata.
MR wykazuje obustronne zmiany demielinizacyjne w istocie białej, z obwódką zapalną, którą można uwidocznić po podaniu środka kontrastowego lub w sekwencji DWI. Diagnostyka opiera się na stwierdzeniu podwyższenia stężenia VLCFA w surowicy, zarówno u chorych, jak i nosicieli/nosicielek choroby. W postaciach mózgowych leczenie jest wyłącznie objawowe. Konieczna jest również suplementacja hormonalna w niedoczynności kory nadnerczy. Olej Lorenza, który obniża stężenie VLCFA we krwi, nadal bywa stosowany, zwłaszcza w okresie przedobjawowym. Nie ma jednak wpływu na wewnątrzkomórkowy metabolizm, więc jest nieskuteczny.
Badania genetyczne potwierdzają chorobę zwłaszcza w przypadku niejednoznacznych wyników biochemicznych. 7, 11, 12, 13 Ponadto umożliwiają poradnictwo genetyczne i ewentualne zaproponowanie leczenia za pomocą przeszczepienia szpiku nosicielowi mutacji niemającemu jeszcze objawów. W badaniach wieloośrodkowych nie stwierdzono korelacji genotyp-fenotyp nawet w obrębie jednej rodziny z X-ALD. 7
Leukodystrofia z zajęciem pnia mózgu i rdzenia oraz podwyższeniem stężenia mleczanów (LBSL)
Leukodystrofia z zajęciem pnia mózgu i rdzenia oraz podwyższeniem stężenia mleczanów w zajętej tkance (LBSL – leukodystrophy with brain stem and spinal cord involvement and lactate elevation) to bardzo rzadka, dziedziczona autosomalnie recesywnie choroba mitochondrialna uwarunkowana mutacjami genu DARS2. W klasycznej postaci przebieg jest najczęściej powolny. Opisano również ciężkie encefalopatie noworodkowe z hipomielinizacją, wykryte dzięki badaniom z zastosowaniem całoeksomowego sekwencjonowania.
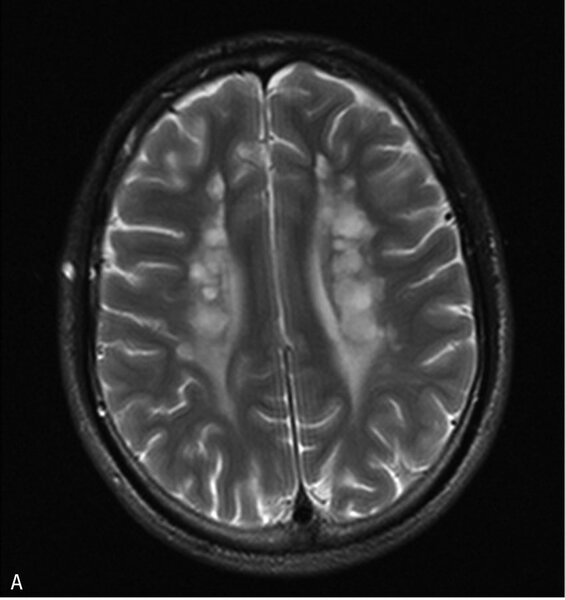
Rycina 5. Leukodystrofia z zajęciem pnia mózgu i rdzenia oraz podwyższonym stężeniem mleczanu w tkance (LBSL). A. W sekwencji T2 w głębokiej istocie białej półkul widoczne okrągłe i owalne ogniska demielinizacji. B. Zajęte sznury tylne rdzenia
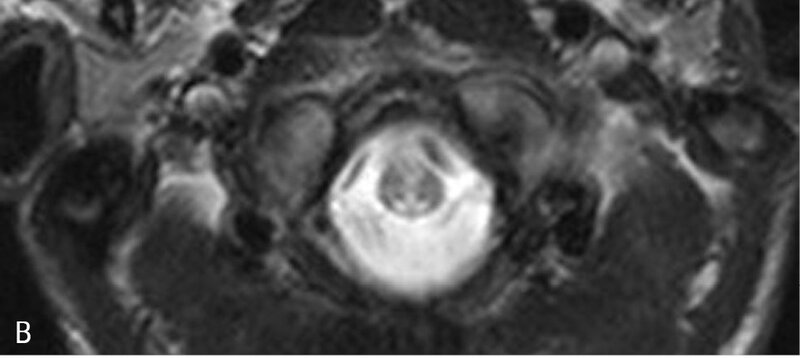
Rycina 5. Leukodystrofia z zajęciem pnia mózgu i rdzenia oraz podwyższonym stężeniem mleczanu w tkance (LBSL). A. W sekwencji T2 w głębokiej istocie białej półkul widoczne okrągłe i owalne ogniska demielinizacji. B. Zajęte sznury tylne rdzenia
LBSL objawia się postępującą ataksją, zespołem pozapiramidowo-piramidowym z przeważającym zajęciem kończyn dolnych i chodem bocianim na skutek zajęcia sznurów tylnych rdzenia. Rozwój umysłowy jest najczęściej prawidłowy. Dość charakterystyczny jest obraz MR, który ukazuje liczne okrągłe ogniska w głębokiej istocie białej mózgu, hiperintensywne w sekwencji T2, hipointensywne w T1; w rdzeniu stwierdza się hiperintensywny sygnał T2 w sznurach tylnych (ryc. 5). W MRS obserwuje się w ich obrębie „pik” mleczanów. Podstawą diagnostyki są badania neuroobrazowe i genetyczne. 7, 9, 10
Leukodystrofia ze znikającą istotą białą (VWM)
VWM jest rzadką chorobą dziedziczoną autosomalnie recesywnie, wywołaną mutacjami w genach pięcioskładnikowego kompleksu EIF2B inicjującego translację. Najczęściej ma powolny przebieg. U dzieci rozwija się na podłożu hipomielinizacji i opóźnionej mielinizacji. Z czasem dochodzi do postępującej demielinizacji z bezodczynowym ubywaniem osłonek mielinowych i pozornym zagęszczeniem oligodendrocytów w obrazie neuropatologicznym. Rozwój umysłowy jest zwykle prawidłowy lub upośledzony w stopniu lekkim. Choroba może ujawnić się ostro stanem podśpiączkowym lub śpiączką podczas infekcji z gorączką, po silnym stresie emocjonalnym lub urazie głowy. Wraz z częściową poprawą pojawiają się regres psychoruchowy oraz ataksja i inne objawy neurologiczne.
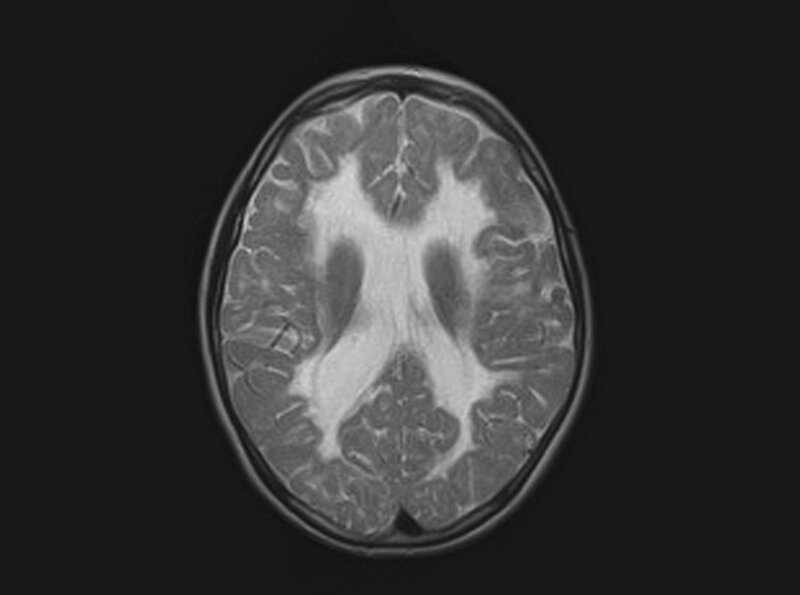
Rycina 6. Leukodystrofia ze znikającą istotą białą (VWM). W sekwencji T2 obustronne zajęcie istoty białej, której sygnał jest zbliżony do sygnału płynu mózgowo-rdzeniowego. Dziewczynka 13-letnia z zespołem pozapiramidowo-móżdżkowym, chorująca od 6 r.ż.
V
WM może również przebiegać przewlekle, z powoli pojawiającą się ataksją, zespołem pozapiramidowo-piramidowym i uogólnioną wiotkością – dzieje się to w ciągu lat, a nawet dekad. Odruchy głębokie mogą być osłabione. Objaw Babińskiego obserwuje się niestale i jest to objaw późny. U dorosłych VWM objawia się ataksją, zespołem piramidowym, powoli narastającymi zaburzeniami poznawczymi, rzadziej psychotycznymi.
Obraz zmian w badaniu MR jest bardzo charakterystyczny – obserwuje się obustronne stopniowe znikanie istoty białej począwszy od okolic okołokomorowych; sygnał istoty białej staje się podobny do sygnału płynu mózgowo-rdzeniowego (ryc. 6). 8, 9, 10 Dorosłe chore z VWM mają niekiedy dysplazję jajników i wymagają hormonalnej terapii zastępczej. Taką postać choroby nazwano ovarioleukodystrofią. W Europie i Polsce najczęstsze są mutacje w genie EIF2B5; u jednej z dużych polskich rodzin z Podhala wykryto mutacje genu EIF2B2.
Choroba Alexandra (ALXDRD)
ALXDRD to dziedziczona autosomalnie dominująco, zwykle sporadyczna LD wywołana mutacjami genu GFAP. Spowodowana jest genetycznie uwarunkowaną nieprawidłową funkcją głównych komórek makrogleju – astrocytów, sensu stricto należy więc do astrocytopatii. Patologicznie zmienione białko GFAP odkłada się w cytoplazmie i wypustkach astrogleju jako tzw. włókna Rosenthala (RF – Rosenthal fibers). Masywne spichrzanie tych włókien obserwuje się głównie okołonaczyniowo, okołokomorowo i podwyściółkowo, ale też w całym mózgu. Prowadzi to do zwiększenia masy i rozmiarów mózgu, co skutkuje wielkogłowiem. 12
U dzieci, u których ALXDRD rozwija się na podłożu hipomielinizacji, można ją łatwo rozpoznać na podstawie charakterystycznego badania MR – przeważającego zajęcia istoty białej płatów czołowych mózgu (ryc. 7). Zależnie od momentu ujawnienia się choroby obserwuje się wielkogłowie prawdziwe (postać wrodzona i wczesnodziecięca) lub względne w stosunku do stwierdzanego zawsze niedoboru wzrostu. ALXDRD postępuje zwykle powoli.
Dzieci z postacią wrodzoną są głęboko upośledzone psychoruchowo i mogą dodatkowo mieć wodogłowie nadnamiotowe wskutek zwężenia wodociągu. U chorych z postacią dziecięcą obserwuje się powoli postępujący zespół spastyczny z przewagą zajęcia kończyn dolnych oraz padaczkę. Rozwój umysłowy jest upośledzony w stopniu lekkim lub umiarkowanym. Chorzy z postacią młodzieńczą, ujawniającą się najczęściej na przełomie 1 i 2 dekady życia, rozwijają się prawidłowo, mają skłonność do krztuszenia się i porannych wymiotów. Może występować niezgrabność ruchowa; niekiedy badanie neurologiczne nie wykazuje odchyleń od stanu prawidłowego. MR mózgowia może ujawnić charakterystyczne zmiany jeszcze w okresie przedobjawowym, przy okazji badań wykonywanych z innej przyczyny, np. w ramach diagnostyki padaczki lub w celu wykluczenia guza. Stopniowo, niekiedy dopiero po kilku kolejnych latach, rozwija się u nich zespół pozapiramidowo-piramidowy. 11, 12 U kilkorga polskich chorych przez wiele lat podejrzewano mózgowe porażenie dziecięce lub niepostępującą encefalopatię z padaczką.
U dorosłych ALXDRD może się objawiać paraparezą spastyczną, otępieniem, zobojętnieniem lub depresją. Neuroobrazowanie nie jest charakterystyczne dla tej postaci choroby, ponieważ zwykle wykazuje niesymetrycznie położone ogniskowe zmiany demielinizacyjne w mózgowiu, w tym w pniu mózgu i istocie białej móżdżku, co może sugerować stwardnienie rozsiane lub zapalenie mózgu. Choroba może przebiegać z okresami poprawy i remisji, co dodatkowo utrudnia rozpoznanie. W tych postaciach konieczna jest diagnostyka molekularna. 1, 3, 7
Choroba Canavan (CD)
CD to dziedziczona autosomalnie recesywnie LDD z wielkogłowiem, wywołana mutacjami genu ASPA. Jej częstość ocenia się na 1/100 000; częściej występuje wśród Żydów aszkenazyjskich. Charakteryzuje się rozległym zgąbczeniem istoty białej z jej wakuolizacją na skutek rozszczepienia blaszek mieliny. Choroba ujawnia się zazwyczaj w niemowlęctwie lub we wczesnym dzieciństwie – występują wielkogłowie i wiotkość, później obserwuje się regres rozwoju psychoruchowego i rozwój zespołu pozapiramidowo-piramidowego z następowym zespołem opuszkowym. Może wystąpić padaczka. Przebieg jest postępujący, chorzy są całkowicie niesprawni ruchowo, mogą doświadczać napadów padaczkowych; względnie długo zachowany jest jedynie kontakt emocjonalny.
Obraz MR wykazuje zajęcie całej istoty białej, łącznie z obszarami podkorowymi, od których proces chorobowy wyraźnie się zaczyna. Zmieniony jest również sygnał z gałek bladych i wzgórz (ryc. 8). W MRS widoczny jest „pik” NAA. Potwierdzenie rozpoznania stanowi wykrycie NAA w moczu w badaniu GCMS, ewentualnie badanie genetyczne. 7, 11, 12, 13
Leukodystrofia z wielkogłowiem i obecnością torbieli podkorowych (MLC)
Leukodystrofia z wielkogłowiem i obecnością torbieli podkorowych (MLC – megalencephalic leukoencephalopathy with a subcortical cysts) jest dziedziczoną autosomalnie recesywnie LDD, rozwijającą się na podłożu hipomielinizacji/dysmielinizacji. Powodują ją mutacje w genach MLC1 lub HEPACAM.
Szybki przyrost obwodu głowy obserwuje się już w 1 r.ż.; w 2 dekadzie dochodzi on do 62-63 cm (>97 centyla). W pierwszych latach życia rozwój psychoruchowy dziecka może być nieznacznie opóźniony lub prawidłowy. Początek choroby obserwuje się po urazach głowy. Pojawia się ataksja, a następnie powolny regres umysłowy. Narastający zespół piramidowo-pozapiramidowy rozwija się po latach; kontakt społeczny i emocjonalny jest długo zachowany. 3, 12
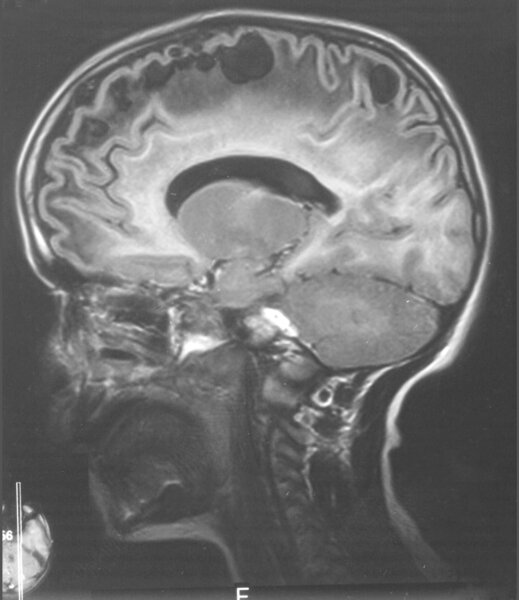
Rycina 9. Leukodystrofia z wielkogłowiem i obecnością torbieli podkorowych (MLC). MR, sekwencja T2, przekrój strzałkowy dla uwidocznienia torbieli podkorowych. 13-letnia dziewczynka z powoli postępującym (od 6 r.ż.) zespołem pozapiramidowo-piramidowo-móżdżkowym, który pojawił się po urazie głowy
MR mózgu ukazuje rozległe zajęcie istoty białej, typowe dla hipomielinizacji/dysmielinizacji z zajęciem włókien podkorowych, a po latach dochodzi do rozległej demielinizacji oraz pojawienia się torbieli podkorowych w płatach czołowych i skroniowych (ryc. 9). Neuropatologicznie stwierdza się rozległe zgąbczenie istoty białej. 5
Leukodystrofia demielinizacyjna dominująca o początku w wieku dorosłym (ADLD)
Leukodystrofia demielinizacyjna dominująca o początku w wieku dorosłym (ADLD – adult-onset autosomal dominant leukodystrophy) jest spowodowana duplikacją genu LMNB1. Ujawnia się w 4-5 dekadzie życia. Cechuje się powolnym przebiegiem, nasilonymi zaburzeniami autonomicznymi (hipotonia ortostatyczna, zaburzenia zwieraczy, takie jak nietrzymanie moczu, zaparcia) oraz postępującą paraparezą spastyczną. Sprawność umysłowa jest długo zachowana. MR wykazuje symetryczne zajęcie głębokiej istoty białej półkul mózgowych. 1, 2, 3, 4, 7
Leukodystrofia z obecnością sferoidów aksonalnych i pigmentacją gleju (HDLS)
Leukodystrofia z obecnością sferoidów aksonalnych i pigmentacją gleju (HDLS – hereditary diffuse leukoencephalopathy with spheroids) to kolejna dziedziczona autosomalnie dominująco LD występująca w wieku dorosłym. Wywołuje ją mutacja genu CSF1R. Ujawnia się w 5 lub 6 dekadzie życia. W obrazie klinicznym dominują otępienie, apraksja oraz parkinsonizm ze sztywnością i bradykinezją, zobojętnienie lub depresja, a z czasem zespół piramidowy. Choroba ma szybki, niepomyślny przebieg, najczęściej w ciągu ok. 6 lat prowadzący do zgonu.
W badaniu MR mózgowia widoczne jest obustronne, nieco asymetryczne zajęcie istoty białej półkul mózgu z przewagą okolic czołowych i ciemieniowych, a także zanik tych płatów. Zmiany dotyczą głębokiej i podkorowej istoty białej. Ich sygnał jest typowy dla demielinizacji. W badaniu neuropatologicznym stwierdzano ubytek osłonek mielinowych, gliozę, głównie podkorową, z odkładaniem się w tych komórkach barwnika zawierającego żelazo oraz sferoidy aksonalne. 5
W piśmiennictwie istnieją opisy kilkupokoleniowych rodzin dotkniętych HDLS. Z racji nasilonych zaburzeń poznawczych choroba ta wymaga różnicowania z otępieniem czołowo-skroniowym, chorobą Alzheimera, a z powodu współistnienia objawów neurologicznych – z chorobą Creutzfeldta-Jakoba i zespołem CADASIL. 3, 4, 7
Genetycznie uwarunkowane leukoencefalopatie (gLE)
Grupa gLE obejmuje choroby, w których uszkodzenie istoty białej wynika zazwyczaj z wrodzonych błędów metabolicznych – ogólnoustrojowych lub dotyczących układu nerwowego – oraz z innych zaburzeń molekularnych (np. niektóre kolagenopatie, choroba Lowe’a, niektóre dystrofinopatie). Przyczyną może być również patologia drobnych naczyń (np. CADASIL, CARASIL i inne). Wśród zaburzeń metabolicznych uszkodzenie istoty białej obserwuje się w:
- aminoacydopatiach (np. nieleczonej fenyloketonurii)
- niektórych kwasicach organicznych (np. kwasicy glutarowej typu 1)
- deficycie receptora folianowego
- galaktozemii
- chorobach lizosomalnych rozwijających się w okresie niemowlęcym (np. chorobie ze spichrzaniem kwasu sjalowego, gangliozydozach GM1 i GM2, ceroidolipofuscynozach neuronalnych, chorobie Niemanna-Picka typu C)
- w innych rzadko występujących błędach metabolicznych, takich jak choroba Menkesa czy deficyt liazy adenylobursztynianowej. 3, 4, 5, 11
Mózgowa dominująca arteriopatia z udarami podkorowymi, leukoencefalopatią i otępieniem (CADASIL)
CADASIL to występująca rodzinnie, powoli postępująca choroba spowodowana mutacjami w jednym allelu genu NOTCH3, ujawniająca się począwszy od 3 dekady życia. Klinicznie obserwuje się napady migreny i/lub przemijającego niedokrwienia mózgu, a następnie postępujące otępienie oraz objawy neurologiczne wynikające z udarów niedokrwiennych, ponadto nietrzymanie moczu.
W badaniu MR w sekwencji T2 widoczne są hiperintensywne, drobne odnaczyniowe ogniska udarowe w istocie białej, które z czasem się ze sobą zlewają, oraz liczne drobne zmiany okołonaczyniowe w zwojach podstawy (stan sitowaty). Diagnostyka opiera się na obrazie MR oraz ewentualnej biopsji skórno-mięśniowej, w której w badaniu mikroskopowo-elektronowym w drobnych tętniczkach wykrywa się złogi elektronowo gęste (GOM – granular osmiophilic material). Aby potwierdzić rozpoznanie, poleca się badanie genetyczne – jako nieinwazyjne. 1, 3, 7, 9
Deficyt liazy adenylobursztynianowej (ADSL)
Deficyt liazy adenylobursztynianowej (ADSL – adenylosuccinate lyase) to bardzo rzadka, dziedziczona autosomalnie recesywnie encefalopatia z upośledzeniem rozwoju, oporną na leczenie padaczką i postępującym zespołem pozapiramidowo-piramidowym. Jej przyczynę stanowi mutacja genu ADSL kodującego enzym biorący udział w syntezie puryn. Nieprawidłowy rozwój psychoruchowy i najczęściej oporne na leczenie drgawki obserwuje się od urodzenia.
Rycina 10. Leukoencefalopatia wskutek niedoboru liazy adenylobursztynianowej (ADSL). MR, sekwencja T2, w przekroju strzałkowym widoczne zajęcie całej istoty białej półkul mózgu z przewagą w okolicach tylnych. Widoczne też uszkodzenie jąder zębatych móżdżku. Dziewczynka 2-miesięczna z lekooporną padaczką od okresu noworodkowego i postępującym zanikiem mózgowia. W wieku 4 miesięcy dziecko zmarło
Obraz MR mózgu wykazuje postępujące uszkodzenie całej istoty białej oraz zaniki korowo-podkorowe; na hipomielinizację nakłada się z czasem demielinizacja, która może się ujawnić po nagłym pogorszeniu stanu ogólnego dziecka (ryc. 10). Chorobę można wykryć na podstawie obecności dwóch metabolitów przemiany puryn w moczu i płynie mózgowo-rdzeniowym: bursztynyloadenozyny (s-Ado – succinyladenosine) i rybozydu bursztynyloaminoimidazolokarboksamidu (SAICAr – succinylaminoimidazole carboxamide riboside). Neuropatologicznie widoczne jest uszkodzenie wszystkich elementów tkanki mózgowej – zarówno neuronów wraz z aksonami, jak i astrogleju oraz oligodendrogleju. W strukturach zmielinizowanych widać rozpad osłonek mielinowych. Przypuszcza się, że przyczynę uszkodzeń stanowi toksyczne działanie metabolitów przemian pośrednich puryn. 3, 13 Diagnostyka genetyczna jest niezbędna w poradnictwie genetycznym.
Leczenie i postępowanie we wrodzonych chorobach istoty białej
W większości LD i gLE możliwe jest jedynie leczenie objawowe i wspierające (rehabilitacja zapobiegająca przykurczom, terapia zajęciowa, prawidłowe odżywianie drogą przezskórnej gastrostomii). W okresie przedobjawowym MLD, GLD oraz X-ALD skuteczne okazało się allogeniczne przeszczepienie szpiku. Z tego leczenia może skorzystać jedynie rodzeństwo probanta (pierwszego chorego w rodzinie) obciążone defektem genetycznym, ponieważ procedury związane z przygotowaniem do przeszczepienia są zbyt obciążające dla chorego z uszkodzeniem OUN. Ze względu na ryzyko choroby przeszczep przeciw gospodarzowi konieczne jest też bardzo staranne dobranie pod względem zgodności tkankowej.
Ostatnio prowadzone są badania nad terapią genową m.in. w X-ALD z zastosowaniem lentiwirusów jako wektorów wprowadzających prawidłowy gen. Olej Lorenza normalizujący jedynie stężenie VLCFA (C22-C26) w surowicy nie jest skuteczny, ponieważ nie ma wpływu na wewnątrzkomórkowy metabolizm. W pozostałych LD, takich jak np. VWM oraz LD o podłożu mitochondrialnym, ważne jest zapobieganie czynnikom mogącym prowadzić do dramatycznych pogorszeń, jak np. urazy głowy, gorączka czy silny stres emocjonalny. 11, 15
Już po napisaniu tej pracy pojawił się wartościowy artykuł Vanderver 16 na temat leukodystrofii u dorosłych. Autorka podaje w nim m.in., które LD mogą rozpocząć się zmianami ogniskowymi w obrazie MR.
Abstract
Leukodystrophies
Leukodystrophies (LD) are genetically determined disorders characterized by the prevalent involvement of central nervous system white matter. The number of related disease entities is estimated at around 90-100. Some authors argue that LDs are disorders which result only from the pathology of glial cells and they also distinguish genetic leukoencephalopathies (gLE) in which white matter damage is secondary to the involvement of other brain tissue components such as blood vessels or to inherited metabolic disorders. The clinical course of LDs is mostly progressive but it may also be static or even improve with time. Most leukodystrophies are often fatal and no curative treatment is known. A distinction is made between hypomyelinating and demyelinating leukodystrophies which have different clinical and MRI pictures. MRI pattern recognition and next-generation sequencing help to establish an accurate diagnosis which is necessary for proper, most frequently symptomatic treatment and for genetic counselling. The paper presents some of the most frequent LDs.
- 1. Van der Knaap MS, Bugiani M. Leukodystrophies: a proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol 2017;134(3):351-82.
- 2. Kevelam SH, Steenweg ME, Srivastava S, et al. Update on Leukodystrophies: A Historical Perspective and Adapted Definition. Neuropediatrics 2016;47(6):349-54.
- 3. Vanderver A, Tonduti D, Schiffmann R, et al. Leukodystrophy overview. In: Pagon RA, Adam MP, Ardinger HH (eds), et al. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016.
- 4. Vanderver A, Prust M, Tonduti D, et al. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol Genet Metab 2015;114(4): 494-500.
- 5. Harding BN, Surtees RAH. Leukoencephalopathy: Metabolic and neurodegenerative diseases of childhood. In: Love S, Louis D, Ellison DW (eds). Greenfield’s neuropathology. London: Hodder Arnold Publication, 2008.
- 6. Vanderver A, Simons C, Helman G, et al. Whole exome sequencing in patients with white matter abnormalities. Ann Neurol 2016;79:1031-7.
- 7. Online Mendelian Inheritance of Man (OMIM); www.ncbi.nlm.nih.gov/omim.
- 8. Schiffmann R, van der Knaap MS. Invited article: An MRI-based approach to the diagnosis of white matter disorders. Neurology 2009;72(8):750-9.
- 9. Van der Knaap MS, Valk J. Magnetic Resonance of Myelination and Myelin Disorders. Berlin: Springer, 2005.
- 10. Steenweg ME, Vanderver A, Blaser S, et al. Magnetic resonance imaging pattern recognition in hypomyelinating disorders. Brain 2010;133(10):2971-82.
- 11. Saudubray JM, Charpentier C. Clinical phenotypes: diagnosis/algorithms. In: Scriver CR, Beaudet AL, Sly W, et al. (eds). The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill, 1995.
- 12. Lyon G, Kolodny EH, Pastores GM. Neurology of Hereditary Metabolic Disease of Children. New York: McGraw-Hill, 2006.
- 13. Zschocke J, Hoffmann GF. Vademecum metabolicum: diagnosis and treatment of inborn errors of metabolism. Friedsriechsdorf: Milupa Metabolics, 2011.
- 14. Parikh S, Bernard G, Leventer RJ, et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol Genet Metab 2015;114(4):501-15.
- 15. Helman G, Van Haren K, Bonkowsky JL, et al. Disease specific therapies in leukodystrophies and leukoencephalopathies. Mol Genet Metab 2015;114(4):527-36.
- 16. Vanderver A. Genetic Leukoencephalopathies in Adults. Continuum (Minneap Minn) 2016;22(3):916-42.
Następny artykuł:

 Dodaj do ulubionych
Dodaj do ulubionych
