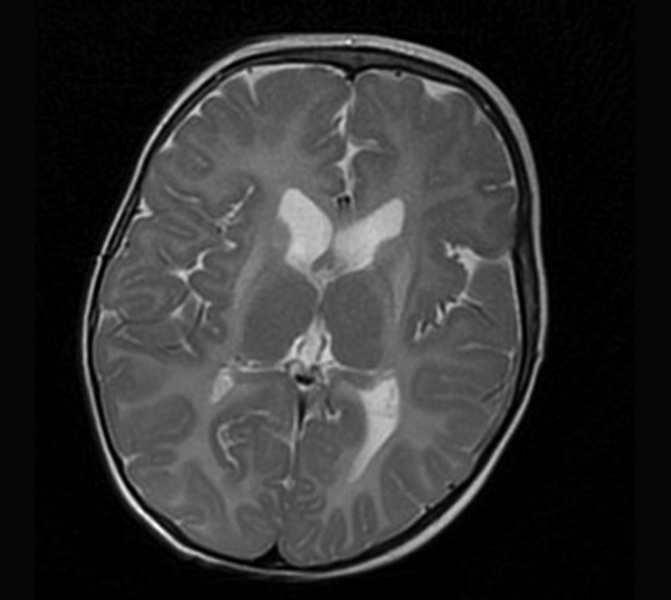
PMD to najczęstsza i najlepiej poznana leukodystrofia hipomielinizacyjna (HLD1). Częstość jej występowania ocenia się na 1-2/100 000 urodzeń. Spowodowana jest mutacją genu PLP1 i dziedziczona recesywnie w sprzężeniu z chromosomem X. Wspomniany gen koduje białko PLP1 oraz izoformę DM20. PMD dotyka najczęściej chłopców, u których może mieć postać wrodzoną, klasyczną lub SPG2. Ta ostatnia może występować u kobiet nosicielek mutacji. W badaniu MR mózgowia stwierdza się rozległą hipomielinizację istoty białej mózgu z zajęciem torebek wewnętrznych, pnia mózgu i móżdżku (ryc. 1).
We wrodzonej postaci PMD, niezwykle rzadkiej, obserwuje się hipotrofię wewnątrzmaciczną, znaczne opóźnienie rozwoju psychoruchowego i fizycznego, skrajną wiotkość, w tym stridor, wrodzone małogłowie oraz oczopląs; odruchy głębokie są zachowane. Szybko dochodzi do niewydolności oddechowej i zgonu.
W klasycznej postaci PMD od okresu niemowlęcego obserwuje się oczopląs, uogólnioną wiotkość oraz opóźnienie rozwoju, zwłaszcza ruchowego. Ponadto występują ataksja móżdżkowa oraz dyskinezy, z czasem dołącza się dystonia. Odruchy głębokie są często wygórowane. Ze względu na powolny przebieg i poprawę niektórych parametrów rozwojowych u wielu chorych z klasyczną postacią PMD może być błędnie rozpoznawana postać móżdżkowa lub pozapiramidowa mózgowego porażenia dziecięcego. W tej postaci dopiero po kilku latach dochodzi do spastyczności. Rozwój umysłowy jest różny: od upośledzenia głębokiego do prawidłowego. W badaniu okulistycznym stwierdza się blade tarcze nerwów wzrokowych, jednak widzenie jest zachowane względnie długo. Najczęstszy typ patogennej mutacji stanowi duplikacja całego genu PLP1. W badaniach z ostatnich lat nie potwierdzono korelacji fenotyp-genotyp – mutacje punktowe i duplikacje całego genu PLP1 wykryto u podłoża ciężkich wrodzonych postaci PMD, ale też względnie łagodnych klasycznych, w tym u chorych z prawidłowym rozwojem umysłowym.7
Leukodystrofia z hipomielinizacją oraz zanikiem zwojów podstawy i móżdżku (HLD6, H-ABC)
Leukodystrofia z hipomielinizacją oraz zanikiem zwojów podstawy i móżdżku (HLD6; H-ABC – hypomyelination with atrophy of basal ganglia and cerebellum) to niezwykle rzadka, sporadycznie występująca choroba wywołana dominującymi mutacjami w jednym allelu genu TUBB4.
W postaci wrodzonej ruchy płodów relacjonowane są jako osłabione; może również występować wielowodzie. Dzieci rodzą się z prawidłową masą i obwodem głowy, jednak przyrost obwodu głowy jest spowolniony i rozwija się małogłowie. Oczopląs, wiotkość oraz opóźnienie psychoruchowe obserwuje się od pierwszych miesięcy życia. Z czasem pojawia się narastający zespół piramidowo-pozapiramidowy ze spastycznością, dystonią, rzadziej z choreoatetozą. Odruchy głębokie są wygórowane. Niekiedy może wystąpić padaczka. W postaciach o późniejszym przebiegu często rozpoznawana jest błędnie postać piramidowo-pozapiramidowa mózgowego porażenia dziecięcego. Rozwój umysłowy jest najczęściej nieprawidłowy.
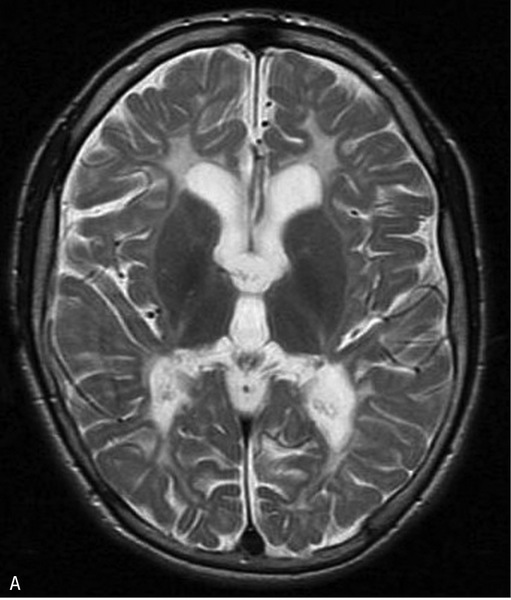
Rycina 2. Leukodystrofia hipomielinizacyjna z zanikiem zwojów podstawy i móżdżku (HLD6). MR, sekwencja T2. Rozległa hipomielinizacja oraz hiperintensywny sygnał zanikających, obkurczonych skorup. Dziewczynka 14-miesięczna z zespołem piramidowo-pozapiramidowym
Na rozpoznanie pozwala badanie MR, w którym oprócz rozległej hipomielinizacji stwierdza się postępujący zanik prążkowia (skorup i głów jąder ogoniastych), a z czasem również móżdżku (ryc. 2). Opisano postacie HLD6 wyłącznie z LD, bez zaniku zwojów podstawy. Choroba ma charakter powoli postępujący i zależnie od ciężkości przebiegu w 2-3 dekadzie życia dochodzi do zgonu.3,4,6
Leukodystrofie hipomielinizacyjne typu 7 i 8
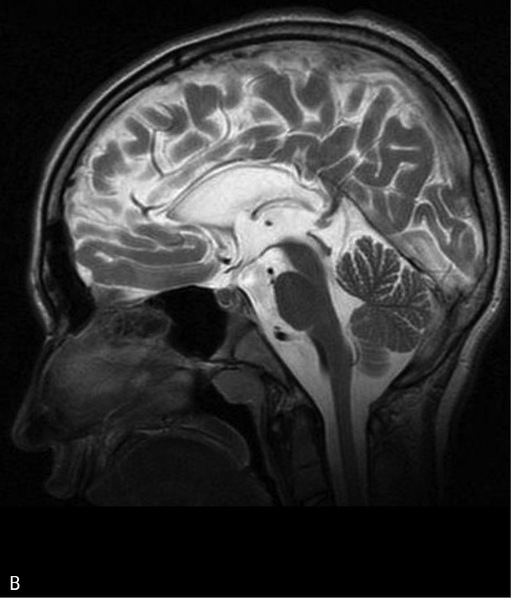
Rycina 3. Leukodystrofia hipomielinizacyjna typu 7 (HLD7) z hipodoncją i hipogonadyzmem hipogonadotropowym. A. MR, sekwencja T2. Hipomielinizacja oraz zmniejszona objętość całej istoty białej półkul mózgu. B. Ścieńczałe ciało modzelowate, widoczny również zanik móżdżku. Chłopiec 10-letni z powoli postępującym zespołem pozapiramidowo-piramidowym i ataksją
Leukodystrofie hipomielinizacyjne typu 7 i 8 (HLD7, HLD8) są związane z mutacjami w genach POLR3A i POLR3B, kodujących polimerazy RNA. Pełną postać określa się mianem zespołu 4H, ponieważ oprócz rozległej hipomielinizacji mózgowia stwierdza się hipodoncję (brak zawiązków niektórych zębów) oraz hipogonadyzm hipogonadotropowy. U wszystkich chorych po kilku latach dochodzi do zmniejszenia objętości ciała modzelowatego oraz zaniku móżdżku. Opisano postacie poronne HLD7/8 wykryte dzięki badaniom genetycznym, bez rozległej hipomielinizacji, jednak u tych chorych w badaniu MR stwierdzano najczęściej zajęcie odnóg tylnych torebek wewnętrznych i/lub zanik móżdżku (ryc. 3). Hipodoncja i zaburzenia hormonalne występowały rzadziej. HLD7/8 mają najczęściej powolny, postępujący przebieg, jednak z reguły im wcześniejsze ujawnienie choroby, tym ten przebieg jest cięższy.
Klinicznie w zespole 4H obserwuje się różnego stopnia opóźnienie rozwoju ruchowego, niezgrabność ruchową, uogólnioną wiotkość oraz powoli postępującą ataksję. Rozwój umysłowy jest opóźniony w różnym stopniu. Odruchy głębokie są osłabione; dopiero po latach pojawia się objaw Babińskiego. Ząbkowanie jest najczęściej opóźnione, brakuje też niektórych zawiązków zębów mlecznych i stałych. Sporadycznie opisywano ząbkowanie noworodkowe. U chorych z zespołem 4H nie pojawiają się samoistnie cechy dojrzewania płciowego i towarzyszy temu niedobór wzrostu, co może prowadzić do błędnego podejrzenia wielohormonalnej niedoczynności przysadki lub zespołu Turnera u dziewcząt. Zależnie od ciężkości przebiegu choroby narastają zaburzenia neurologiczne i regres umysłowy lub otępienie. W postaciach poronnych choroba może się ujawnić nawet w 2 dekadzie życia w postaci narastających zaburzeń chodu i ataksji, a na rozpoznanie może naprowadzić hipodoncja.16 Leczenie jest objawowe. W postaciach pełnoobjawowych wskazana jest suplementacja hormonalna.
Delecja 18q(-)
Delecja 18q(-) to dysLD związana z brakiem MBP, dla którego gen znajduje się w terminalnym odcinku długiego ramienia chromosomu 18. Obraz kliniczny jest różny w zależności od wielkości delecji i związanego z tym ubytku materiału genetycznego. Obserwuje się upośledzenie psychoruchowe lub niezgrabność ruchową, małogłowie, różne cechy dysmorficzne twarzoczaszki, w tym uszu (łącznie z zarośnięciem przewodu słuchowego i niedosłuchem lub głuchotą), niekiedy wady serca, rzadziej wady kończyn i narządów płciowych. W cięższych postaciach występują oczopląs, ataksja i zespół pozapiramidowo-piramidowy. Stężenie białka w płynie mózgowo-rdzeniowym jest prawidłowe.
Obraz MR mózgu charakteryzuje się słabym zróżnicowaniem sygnału kory i istoty białej z nieostro odgraniczonymi ogniskami o różnej intensywności sygnału, niekiedy jakby rozmytymi w sekwencji T2, FLAIR oraz T1; może występować zanik móżdżku. W diagnostyce niezbędne są badania genetyczne.3,7
Wrodzone choroby istoty białej z demielinizacją
LDD to choroby spowodowane postępującym uszkodzeniem mieliny. Zazwyczaj ich przyczynę stanowią wrodzone błędy metaboliczne.1,3,12,13,15 W Polsce i na świecie najczęstsze są choroby lizosomalne – leukodystrofia metachromatyczna (MLD) i leukodystrofia globoidalna [GLD – globoid cell leukodystrophy]), peroksyzomalne (X-ALD) oraz mózgowe kwasice organiczne (CD). Do tej grupy zaliczają się też postacie ALXDRD i VWM o późnym początku, jak również gLE z uszkodzeniem mieliny wtórnym do innych zaburzeń, niezależnych od dysfunkcji komórek glejowych.
Objawy kliniczne poszczególnych LDD nie są swoiste, a przebieg jest zwykle tym cięższy, im wcześniej te objawy się ujawnią. Po okresie prawidłowego rozwoju u niemowląt i małych dzieci dochodzi do regresu psychoruchowego, u starszych – do zaburzeń chodu i ataksji. Jeśli współistnieje neuropatia obwodowa, obserwuje się przejściową wiotkość. Z czasem, w ciągu kilku lub kilkunastu miesięcy, pojawiają się spastyczny niedowład czterokończynowy, zaburzenia połykania i odmóżdżenie. U dorosłych dochodzi do ataksji, otępienia i spastyczności, a objawy kliniczne postępują w ciągu kolejnych lat.