W płynie mózgowo-rdzeniowym w okresie aktywnego rozpadu mieliny stwierdza się podwyższone stężenie białka. Diagnostyka opiera się na analizie obrazu klinicznego, badaniach neuroobrazowych, które również wymagają precyzyjnej analizy umiejscowienia zmian i ich charakteru, badaniach biochemicznych, elektrofizjologicznych, a ostatnio także na badaniach genetycznych.11,12
Leukodystrofia metachromatyczna (MLD)
MLD to najczęstsza LDD w Polsce i na świecie – częstość jej występowania ocenia się na 1/20 000 urodzeń. Uwarunkowana jest autosomalnie recesywnie mutacjami genu ARSA i należy do chorób lizosomalnych. Obserwuje się pełne spektrum postaci, od niemowlęcych do obserwowanych u dorosłych. Przebieg u niemowląt i małych dzieci jest najczęściej szybki i niepomyślny. Im wcześniej ujawni się choroba, tym cięższy przebieg. Po etapie zatrzymania rozwoju w ciągu kilku tygodni dochodzi do regresu, spastyczności i odmóżdżenia. U dzieci starszych po okresie zaburzeń chodu, ataksji, niekiedy przejściowej wiotkości wskutek neuropatii obwodowej w ciągu kilku miesięcy rozwija się spastyczny niedowład czterokończynowy. U młodzieży i dorosłych choroba przebiega wolniej, a objawom piramidowym towarzyszą ataksja i postępujące otępienie. Występują również postacie z izolowanym otępieniem.
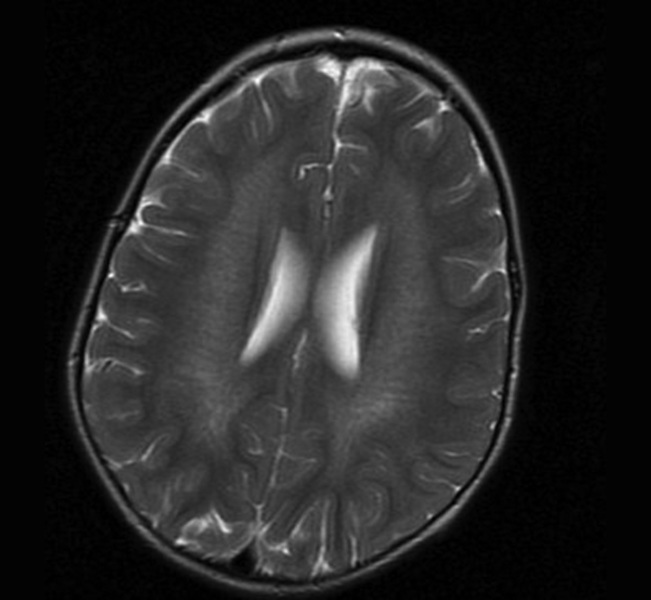
Rycina 4. Leukodystrofia metachromatyczna (MLD) potwierdzona molekularnie. MR, sekwencja T2. W istocie białej głębokiej widoczne jeszcze zachowane włókna nerwowe u 3-letniego chłopca z 3-miesięcznym wywiadem chorobowym i objawami ataksji móżdżkowej
Badania neuroobrazowe ukazują obustronne zmiany demielinizacyjne głębokiej istoty białej. W początkowej fazie choroby widoczne są zachowane włókna nerwowe (ryc. 4). Diagnostyka opiera się na stwierdzeniu znacznie obniżonej aktywności enzymu arylosulfatazy A, a ostatnio coraz częściej na badaniach genetycznych. Obserwuje się korelację genotypowo-fenotypową.3,7
Leukodystrofia globoidalna Krabbego (GLD)
GLD to również dziedziczona autosomalnie recesywnie choroba lizosomalna wynikająca z mutacji genu GACL. W Polsce jest rzadsza niż MLD i występuje z częstością 1/40 000. Może się ujawnić począwszy od okresu niemowlęcego do wieku dorosłego. Przebieg GLD jest tym cięższy, im wcześniej choroba da objawy.
U niemowląt obserwuje się szybki regres psychoruchowy oraz trudny do opanowania niepokój – dzieci sprawiają wrażenie cierpiących, przestają jeść, szybko rozwijają się tetrapareza spastyczna i odmóżdżenie. U dzieci starszych przebieg jest nieco wolniejszy. Po przejściowym okresie zatrzymania rozwoju, ataksji oraz wiotkości wskutek demielinizacyjnej neuropatii obwodowej rozwija się postępujący niedowład spastyczny czterokończynowy, następuje regres rozwoju i w ciągu paru lat dochodzi do zgonu. Postacie typu dorosłe cechują się ataksją, spastycznością z przewagą kończyn dolnych i otępieniem.
W obrazie MR u niemowląt stwierdza się rozlane zmiany w istocie białej zaczynające się w pniu mózgu i móżdżku, tj. w strukturach już zmielinizowanych, a po kilku miesiącach w obu półkulach; sygnał wzgórz jest nieco hiperintensywny; nerwy wzrokowe są pogrubiałe. U starszych dzieci widoczne są rozległe zmiany w istocie białej półkul z zaoszczędzeniem U-włókien, a w postaciach dorosłych – przewaga zajęcia płatów ciemieniowo-potylicznych. Nie obserwuje się korelacji fenotyp-genotyp, w jednej rodzinie mogą występować różne postacie choroby.
Uważa się, że w GLD patogenną rolę odgrywa psychozyna (metabolit cerebrozydów), która uszkadza oligodendrocyty, co prowadzi do demielinizacji. Ostatnio zwrócono też uwagę na inicjującą rozpad mieliny rolę prozapalnych cytokin, wydzielanych przez mikroglej. Komórki globoidalne charakterystyczne dla GLD widoczne są w badaniu mikroskopowym w obszarach rozpadu mieliny. W diagnostyce stosuje się badania biochemiczne (ocena aktywności resztkowej galaktocerebrozydazy) i coraz częściej badania genetyczne.3,7,12
Należy pamiętać, że istnieją rzadkie postacie MLD i GLD, których przebieg jest podobny jak w chorobach klasycznych, uwarunkowane deficytami kofaktorów – sapozyn. Wymagane są celowane badania molekularne w razie silnego podejrzenia tych postaci.7,13
Adrenoleukodystrofia sprzężona z chromosomem X (X-ALD)
X-ALD to druga co do częstości LDD w Polsce. Częstość jej występowania ocenia się na 1/20 000-50 000 urodzeń. X-ALD to choroba peroksyzomalna, uwarunkowana recesywną mutacją genu ABCD1, który koduje białko błony peroksyzomalnej. Jest to transporter przenoszący kwasy tłuszczowe o bardzo długim łańcuchu (VLCFA – very long chain fatty acids) do wnętrza organelli.
Wyróżnia się postaci mózgowe X-ALD, występujące głównie u chłopców w wieku 4-12 lat, rzadziej u młodych mężczyzn, oraz adrenomieloneuropatię, notowaną głównie u dorosłych. Każdej z tych postaci może towarzyszyć niewydolność kory nadnerczy (>60% chorych), która bywa niekiedy jedynym, izolowanym objawem. U dorosłych chorych możliwe są impotencja oraz niewydolność gonad. U kobiet nosicielek mutacji objawy X-ALD mogą być łagodniejsze. Postać mózgowa manifestuje się zaburzeniami chodu, ataksją, niekiedy drgawkami, a następnie szybko postępującym (kilka miesięcy) zespołem porażenia czterokończynowego z wygórowanymi odruchami i spastycznością. Objawom tym towarzyszą często zaburzenia emocjonalne oraz zaburzenia zachowania, rzadziej objawy psychotyczne. Zwykle po kilku miesiącach dochodzi do zespołu odmóżdżenia; średnie przeżycie chorych z postacią mózgową wynosi 2 lata.
MR wykazuje obustronne zmiany demielinizacyjne w istocie białej, z obwódką zapalną, którą można uwidocznić po podaniu środka kontrastowego lub w sekwencji DWI. Diagnostyka opiera się na stwierdzeniu podwyższenia stężenia VLCFA w surowicy, zarówno u chorych, jak i nosicieli/nosicielek choroby. W postaciach mózgowych leczenie jest wyłącznie objawowe. Konieczna jest również suplementacja hormonalna w niedoczynności kory nadnerczy. Olej Lorenza, który obniża stężenie VLCFA we krwi, nadal bywa stosowany, zwłaszcza w okresie przedobjawowym. Nie ma jednak wpływu na wewnątrzkomórkowy metabolizm, więc jest nieskuteczny.
Badania genetyczne potwierdzają chorobę zwłaszcza w przypadku niejednoznacznych wyników biochemicznych.7,11-13 Ponadto umożliwiają poradnictwo genetyczne i ewentualne zaproponowanie leczenia za pomocą przeszczepienia szpiku nosicielowi mutacji niemającemu jeszcze objawów. W badaniach wieloośrodkowych nie stwierdzono korelacji genotyp-fenotyp nawet w obrębie jednej rodziny z X-ALD.7
Leukodystrofia z zajęciem pnia mózgu i rdzenia oraz podwyższeniem stężenia mleczanów (LBSL)
Leukodystrofia z zajęciem pnia mózgu i rdzenia oraz podwyższeniem stężenia mleczanów w zajętej tkance (LBSL – leukodystrophy with brain stem and spinal cord involvement and lactate elevation) to bardzo rzadka, dziedziczona autosomalnie recesywnie choroba mitochondrialna uwarunkowana mutacjami genu DARS2. W klasycznej postaci przebieg jest najczęściej powolny. Opisano również ciężkie encefalopatie noworodkowe z hipomielinizacją, wykryte dzięki badaniom z zastosowaniem całoeksomowego sekwencjonowania.
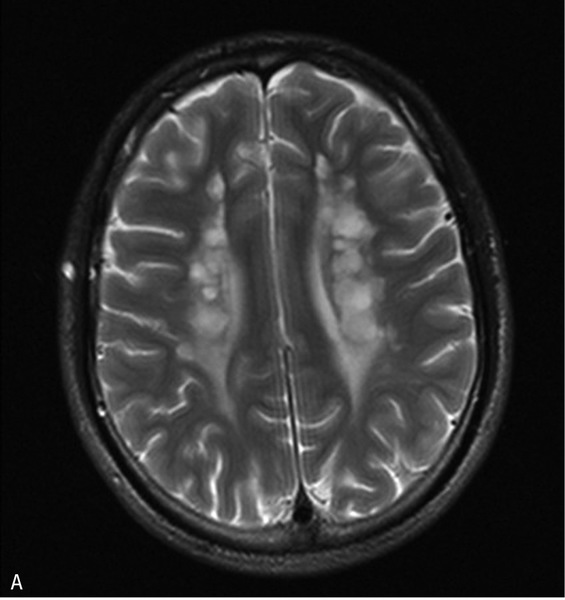
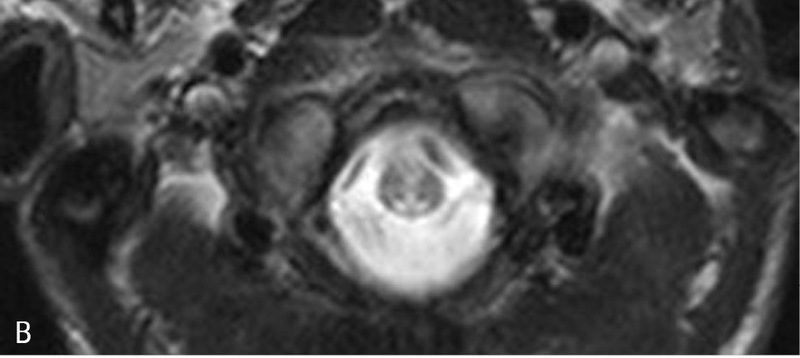
Rycina 5. Leukodystrofia z zajęciem pnia mózgu i rdzenia oraz podwyższonym stężeniem mleczanu w tkance (LBSL). A. W sekwencji T2 w głębokiej istocie białej półkul widoczne okrągłe i owalne ogniska demielinizacji. B. Zajęte sznury tylne rdzenia