Rzadkie choroby ośrodkowego układu nerwowego z zajęciem naczyń mózgu
lek. Dominik Siutka1
dr hab. n. med. Waldemar Brola2

lek. Dominik Siutka

dr hab. n. med. Waldemar Brola
- Szczegółowe omówienie rzadkich mikroangiopatii związanych z mutacjami w genach COL4A1 i COL4A2, NOTCH3, GLA, HTRA1 oraz TREX1 będących przyczynami udarów u osób młodych i udarów o niejasnej etiologii
- Przy niejednoznacznym obrazie radiologicznym udaru konieczne jest rozszerzenie diagnostyki, zwłaszcza jeśli dotyczy on osób młodych bez ewidentnego wywiadu nadciśnienia tętniczego i innych czynników ryzyka, a wywiad rodzinny jest obciążający
W ostatnich latach coraz większą uwagę zwraca się na udary mózgu u osób młodych i udary o niejasnej etiologii. Jednymi z możliwych przyczyn są choroby małych naczyń (SVD – small vessel disease). Te przewlekłe i postępujące schorzenia dotyczą drobnych, przeszywających naczyń krwionośnych mózgu, tzn. małych tętnic, tętniczek, naczyń włosowatych i małych żył zaopatrujących istotę białą i głębokie struktury istoty szarej1.
Choroby małych naczyń są związane z mutacjami w genach COL4A1 i COL4A2, NOTCH3, GLA, HTRA1 oraz TREX1. Są to rzadkie schorzenia objawiające się charakterystycznymi zespołami neurologicznymi, a także specyficznymi zmianami radiologicznymi oraz widocznymi w badaniach neuropatologicznych¹-3. Klinicznie fenotypy choroby małych naczyń różnią się w zależności od postaci. W postaci ostrej objawiają się udarami lakunarnymi bądź głębokimi krwotokami, a w postaci przewlekłej – napadami migreny z aurą, objawami pozapiramidowymi, otępieniem, depresją czy zaburzeniami chodu (tab. 1)3. Ich występowaniu mogą towarzyszyć objawy nieneurologiczne (zmiany skórne, oczne, kostne, zaburzenia hormonalne, nefrologiczne i kardiologiczne).
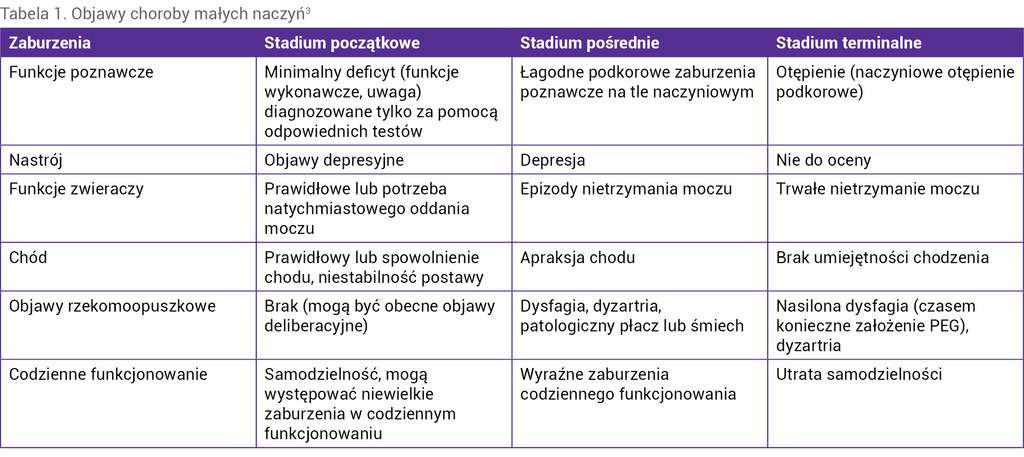
Tabela 1. Objawy choroby małych naczyń3
Przy niejednoznacznym obrazie radiologicznym konieczne jest rozszerzenie diagnostyki, zwłaszcza jeśli udar dotyczy osób młodych bez ewidentnego wywiadu nadciśnienia tętniczego i innych czynników ryzyka, a wywiad rodzinny jest obciążający. Często o ostatecznym rozpoznaniu rozstrzyga badanie genetyczne.
Zespół CADASIL
Zespół CADASIL (mózgowa autosomalna dominująca arteriopatia z podkorowymi zawałami i leukoencefalopatią; cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy) to spowodowana mutacją genu NOTCH3 na chromosomie 19p13.12 genetycznie uwarunkowana choroba małych naczyń mózgowych4. Szacuje się, że CADASIL występuje z częstością 1:25 000-50 000, ale dokładne dane nie są znane ze względu na trudności w rozpoznawaniu choroby. W niektórych izolowanych populacjach może występować zdecydowanie częściej niż w populacji ogólnej3-5. Objawy typowe dla tej choroby to migrena z aurą (20-40%), udar mózgu (40-80%) i zaburzenia poznawcze (100%)3. Migrena z aurą pojawia się w trzeciej dekadzie życia, udar – w czwartej, a zaburzenia poznawcze w szóstej dekadzie życia. Mogą wystąpić również zaburzenia nastroju i apatia (około 20% chorych), padaczka (10%), mikrokrwawienia (31-69%), sporadycznie – udary krwotoczne i objawy pozapiramidowe4. Objawy choroby występują w różnych konfiguracjach i u pacjentów w różnym wieku. Zespół ma bardzo różnorodny obraz kliniczny, nawet wśród członków tej samej rodziny2,3.
Rezonans magnetyczny wykazuje bardzo charakterystyczny obraz zmian istoty białej (zmiany w sekwencji T2 wyprzedzają objawy kliniczne o około 15 lat i są widoczne już około 35 r.ż.). Zwykle umiejscowione są podkorowo i okołokomorowo, szczególnie w płacie skroniowym, torebce zewnętrznej, płatach czołowym i ciemieniowym. Zajęcie przedniej części płata skroniowego (objaw O’Sullivana) i torebki zewnętrznej uważane jest za charakterystyczną radiologiczną cechę choroby3.
W badaniu neuropatologicznym pacjentów cierpiących na CADASIL stwierdza się zmiany typowe dla przewlekłej choroby małych naczyń mózgu, zlokalizowane w istocie białej półkul mózgowych w okolicach okołokomorowych i jądrze półowalnym. Udary lakunarne są umiejscowione nie tylko w istocie białej, lecz także w jądrach podkorowych2,3.
W badaniu histopatologicznym stwierdza się zwyrodnienie i ubytek miocytów gładkich w tętnicach małego i średniego kalibru oraz gromadzenie się w ścianach małych naczyń osmofilnego materiału ziarnistego (GOM – granular osmiophilic material)4. Chociaż objawy choroby wiążą się głównie z układem nerwowym, zmiany GOM obserwuje się też w innych organach: śledzionie, wątrobie, nerkach, mięśniach oraz w skórze. Dzięki łatwemu dostępowi czułość rozpoznania choroby za pomocą biopsji skóry jest wysoka3.
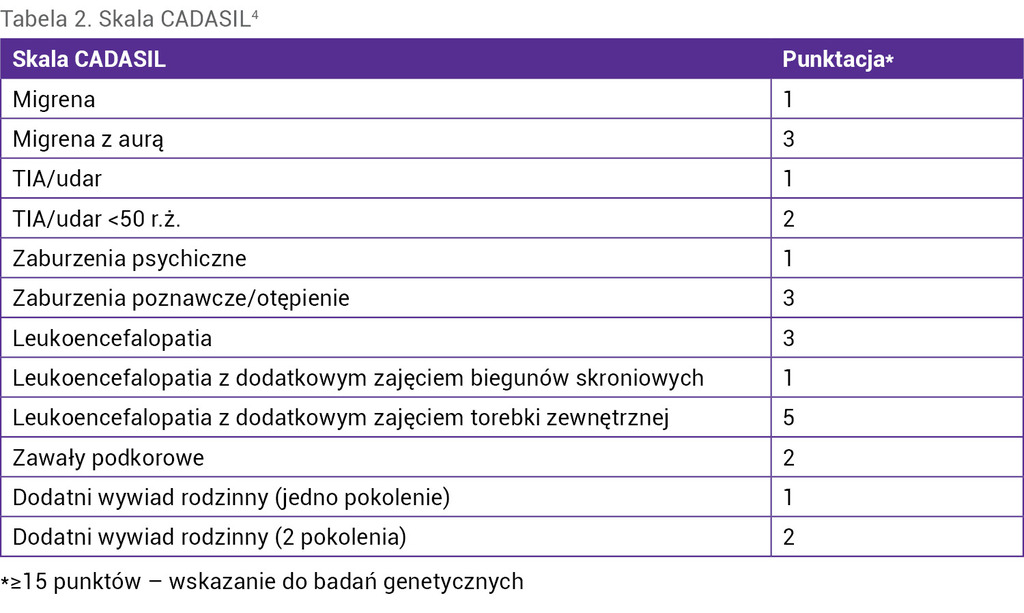
Tabela 2. Skala CADASIL4
Kryteriami umożliwiającymi rozpoznanie choroby są dodatni wynik badania genetycznego oraz obecność złogów GOM w naczyniach w biopsji skóry lub mięśnia szkieletowego. W ustaleniu wskazań do badania genetycznego może być przydatna skala CADASIL (tab. 2)4. Ze względu na nieznany patomechanizm choroby możliwe jest tylko leczenie objawowe, podobne jak w migrenie sporadycznej czy udarze mózgu.
Zespół CARASIL
Podobnym do zespołu CADASIL schorzeniem, również związanym z uszkodzeniem małych naczyń, jest zespół CARASIL (autosomalna recesywna arteriopatia z zawałami podkorowymi i leukoencefalopatią; cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy) występujący głównie w Azji i zwany przez autorów japońskich zespołem Maeda6,7.