Osoby z rozpoznaniem choroby Fabry’ego powinny być poddane kompleksowej wielospecjalistycznej diagnostyce i opiece. Wczesne ustalenie rozpoznania choroby Fabry’ego umożliwia zastosowanie zastępczej terapii enzymatycznej. Obecnie dostępne są dwa preparaty: agalzydaza β i agalzydaza α12. Leczenie jest najskuteczniejsze w pierwszych stadiach choroby, przed uszkodzeniem narządowym. Terapia zmniejsza ból neuropatyczny i poprawia klirens kreatyniny, ale nie ma wpływu na rokowanie. Leczenie przeciwzakrzepowe również nie wpływa na rokowanie w udarze u osób z chorobą Fabry’ego. Postuluje się włączanie terapii zastępczej u chłopców od 7 roku życia pozostających bez objawów12. Dodatkowo stosuje się leczenie objawowe, ukierunkowane na uszkodzone narządy. Duże nadzieje wiąże się obecnie z badaniami skriningowymi noworodków, umożliwiającymi wczesne włączenie enzymatycznej terapii zastępczej12.
Mikroangiopatie związane z mutacjami genów COL4A1 i COL4A2
Mikroangiopatie te są wywołane mutacją w genach COL4A1 (13q34) lub COL4A2 (13q34), które kodują łańcuchy kolagenu IV, budującego błonę podstawną naczyń krwionośnych, struktur oka, kłębuszków nerkowych oraz błonę podstawną mózgu15.
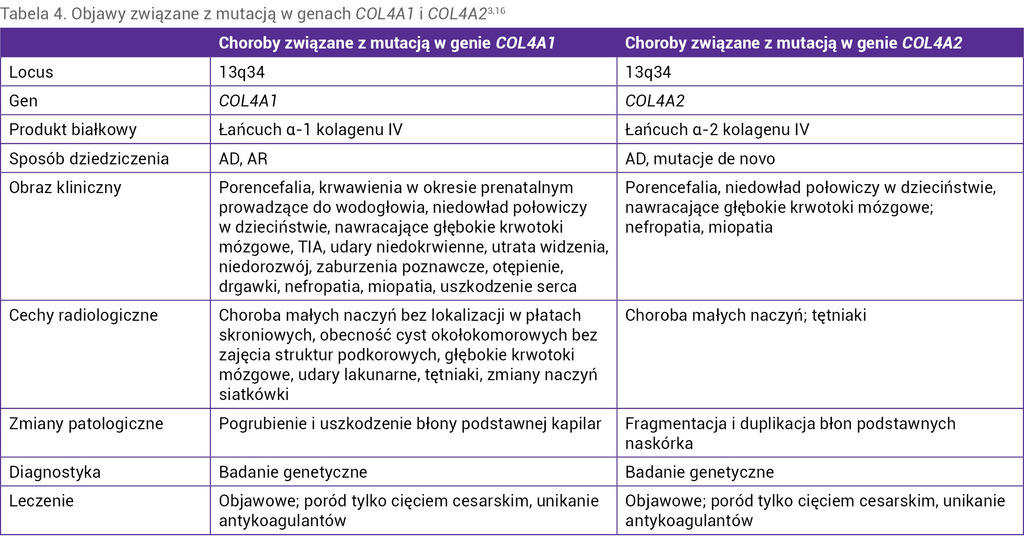
Tabela 4. Objawy związane z mutacją w genach COL4A1 i COL4A23,16
Obraz kliniczny mikroangiopatii charakteryzuje się bardzo szerokim zakresem objawów (tab. 4). Do odchyleń w badaniu neurologicznym zaliczamy niedowłady połowicze, migreny, nieprawidłowy rozwój umysłowy, zaburzenia poznawcze, otępienie, napady padaczkowe. Udary niedokrwienne i krwotoki śródmózgowe występują w każdym wieku, również w okresie prenatalnym lub w trakcie porodu. Na szczególną uwagę zasługują krwawienia, które mogą pojawiać się spontanicznie, w wyniku urazów lub leczenia przeciwkrzepliwego u pacjentów bez nadciśnienia <50 r.ż. Udar mózgu często okazuje się pierwszym objawem choroby15.
Wśród objawów ze strony narządu wzroku wyróżniamy jaskrę, zaćmę, przemijające niedowidzenie spowodowane krwotokami, dysgenezję przedniego odcinka gałki ocznej (anomalia Axenfelda-Riegera). Do innych częstych objawów należy zaliczyć krwiomocz, białkomocz, torbiele nerek, tętniaki tętnicy szyjnej wewnętrznej, objaw Raynauda, nadkomorowe zaburzenia rytmu i skurcze mięśniowe15-17.
W badaniach obrazowych charakterystyczny jest obraz podobny do angiopatii nadciśnieniowej – okołokomorowe intensywne zmiany istoty białej z udarami niedokrwiennymi, krwotocznymi, lakunarnymi i cystami okołokomorowymi16.
W badaniach histopatologicznych naczyń skóry stwierdza się pogrubiałą błonę podstawną naczyń włosowatych oraz fragmentację i duplikację błon podstawnych naskórka16,17. Ostateczne rozpoznanie można ustalić na podstawie sekwencjonowania genów.
Dotychczas udało się wyodrębnić kilka fenotypów klinicznych zaliczanych do zakresu COL4A1:
- porencefalia dziedziczona autosomalnie dominująco typu I (autosomal dominant porencephaly type I)16:
- występuje w okresie noworodkowym i niemowlęcym
- charakteryzuje się widocznymi w MR jamami płynowymi (porencefalicznymi) będącymi pozostałością krwotoków śródmózgowych w życiu płodowym i noworodkowym, mikrokrwotokami, okołokomorową leukoencefalopatią, udarami lakunarnymi i zwapnieniami
- objawia się niedowładami połowiczymi u noworodków, niesprawnością intelektualną, napadami padaczkowymi, dystonią, udarami i migrenami; pierwszym objawem może być udar mózgu u wcześniej pozostającego bez objawów dorosłego
- mózgowa choroba małych naczyń mózgowych przebiegająca z krwotokiem (brain SVD with hemorrhage)17:
- rodzinna leukoencefalopatia naczyniowa zależna od COL4A1
- w MR charakteryzuje się zajęciem małych naczyń mózgowych z rozlaną okołokomorową leukoencefalopatią, zawałami lakunarnymi, mikrokrwawieniami, krwotokami śródmózgowymi, poszerzonymi przestrzeniami okołonaczyniowymi i zwapnieniami
- niedowłady połowicze w okresie niemowlęcym, napady padaczkowe, migreny z aurą, krwotoki śródmózgowe przed 50 r.ż., zaćma wrodzona, kręte naczynia siatkówki, anomalia przedniego odcinka oka typu Axenfelda-Riegera18
- rzadziej występują skurcze mięśni ze wzrostem kinazy kreatynowej, uszkodzenie nerek i niedokrwistość hemolityczna
- choroba małych naczyń mózgowych z anomalią Axenfelda-Riegera (brain SVD with Axenfeld-Rieger Anomaly)18:
- zespół wad wrodzonych charakteryzujący się występowaniem obustronnych wad przedniego odcinka oka skojarzonych z wadami rozwojowymi zębów, środkowej części twarzy i jamy brzusznej
- dziedziczna angiopatia z krętością naczyń siatkówki, nefropatią, tętniakami i kurczami mięśni (HANAC – hereditary angiopathy with nephropathy, aneurysms, and muscle cramps syndrome)19
- choroba małych naczyń z leukoencefalopatią, zawałami lakunarnymi, mikrokrwawieniami i poszerzonymi przestrzeniami okołonaczyniowymi, dodatkowo występują nefropatia, mnogie lub pojedyncze tętniaki tętnic szyjnych, kręte naczynia siatkówki i skurcze mięśniowe; udary niedokrwienne i krwotoczne występują rzadko
- autosomalna dominująca mikroangiopatia mostu z leukoencefalopatią (PADMAL – pontine autosomal dominant microangiopathy with leukencephalopathy)20:
- charakteryzuje się zawałami lakunarnymi mostu oraz uszkodzeniem podkorowej i okołokomorowej istoty białej; uszkodzenie płatów skroniowych i krwawienia występują rzadko
- nie ma leczenia przyczynowego, zapobieganie powikłaniom choroby polega na redukcji czynników ryzyka udarów mózgu (unikanie palenia tytoniu, leczenie nadciśnienia tętniczego), unikaniu leczenia przeciwkrzepliwego i urazów mogących powodować krwotoki16.
Zespół CRMCC
Zespół CRMCC (cerebroretinal microangiopathy with calcifications and cysts), zwany także zespołem „Coats plus”, należy do rzadkich genetycznie uwarunkowanych chorób małych naczyń21. Do cech charakterystycznych zespołu należą przede wszystkim zmiany wewnątrzczaszkowe w postaci zwapnień, torbieli i leukoencefalopatii, z towarzyszącym uszkodzeniem naczyń siatkówki (obecność telangiektazji i wysięków)21. Ponadto do objawów zespołu CRMCC należą: osteopenia z tendencją do złamań, supresja szpiku kostnego, tendencja do krwawień żołądkowo-jelitowych oraz nadciśnienie wrotne. Nazwa zespół „Coats plus” pochodzi od choroby Coatsa, w której obserwuje się izolowaną mikroangiopatię naczyń siatkówki, bez uszkodzeń ze strony innych narządów22. Częstość występowania zespołu szacuje się na <1/100 000.
Najczęstszym objawem zespołu CRMCC są zaburzenia ze strony OUN w postaci drgawek, ataksji, dystonii, zaburzeń ruchowych, a także deficytów poznawczych21. Nieprawidłowo zbudowane naczynia siatkówki predysponują do powstawania wysięków między warstwami siatkówki, co klinicznie objawia się w postaci utraty wzroku. Wraz z wiekiem dochodzi do nasilenia deficytów neurologicznych i postępującego pogorszenia widzenia. Pierwsze objawy pojawiają się we wczesnym dzieciństwie, a nawet już w życiu płodowym. Zazwyczaj obserwuje się opóźniony wzrost i nieprawidłowy rozwój kości. Na skutek osteopenii dochodzi do złamań patologicznych, a supresja szpiku kostnego powoduje niedokrwistość. Osoby z zespołem „Coats plus” charakteryzują się ponadto przedwczesnym siwieniem, nieprawidłową budową włosów i paznokci, a także zaburzoną pigmentacją skóry, mogą występować u nich plamy typu café au lait. Za zagrażające życiu powikłania zespołu uważa się marskość wątroby, nadciśnienie wrotne i poważne krwawienia żołądkowo-jelitowe22.
Przyczyną zespołu CRMCC jest heterozygotyczna mutacja w genie CTC1 (conserved telomere maintenance component 1) znajdującym się na chromosomie 17 (mutacja 17p13.1)22. Gen CTC1 koduje białko wchodzące w skład kompleksu dla replikacji telomerów, które są niezbędne dla zachowania stabilności genomu. W wyniku mutacji tego genu dochodzi do przesunięcia ramki odczytu, co prowadzi do syntezy niestabilnych bądź skróconych białek, niebędących w stanie stworzyć kompleksu z pozostałymi składnikami. W efekcie dochodzi do skrócenia telomerów bądź tworzenia chromosomów fuzalnych22.
Zespół RVCL
Zespół RVCL (autosomal dominant retinal vasculopathy with cerebral leukodystrophy) jest leukodystrofią mózgową z waskulopatią siatkówki dziedziczoną autosomalnie dominująco (chromosom 3p21.1 – p21.3, gen TREX1)24. Do zakresu choroby zaliczamy fenotypy kliniczne:
- waskulopatię mózgowo-siatkówkową (CRV – cerebro-retinal vasculopathy)
- dziedziczną endoteliopatię, retinopatię, nefropatię i udar (HERNS – hereditary endotheliopathy, retinopathy, nephropathy and stroke)
- dziedziczną retinopatię naczyniową (HVR – hereditary vascular retinopathy).
Pierwsze objawy pojawiają się w 40-50 r.ż. Zazwyczaj są to symptomy ze strony narządu wzroku o charakterze postępujących zaburzeń widzenia (waskulopatia naczyń siatkówki, nowotworzenie naczyń w tarczy nerwu wzrokowego, krwotoki do siatkówki, mikrotętniaki plamki żółtej, zanik naczyń włosowatych zaczynający się od plamki żółtej)23. W dalszej kolejności dołączają się objawy neurologiczne: udary niedokrwienne/TIA, migrena, zaburzenia poznawcze, zaburzenia psychiczne (zaburzenia osobowości, depresja, niepokój), drgawki, zgon w ciągu 10 lat od rozpoznania23. Mogą wystąpić również objawy uogólnionego niedokrwienia narządów wewnętrznych, zespół Raynauda, marskość wątroby, niewydolność nerek i martwica kości.
W MR stwierdza się rozlaną okołokomorową leukoencefalopatię i zawały lakunarne związane z zajęciem małych naczyń, czasem dodatkowo pseudotumor w postaci zmiany wzmacniającej się po środku cieniującym zlokalizowanej w mózgu lub móżdżku, otoczonej obrzękiem (podobnej do martwicy po napromieniowaniu)23.