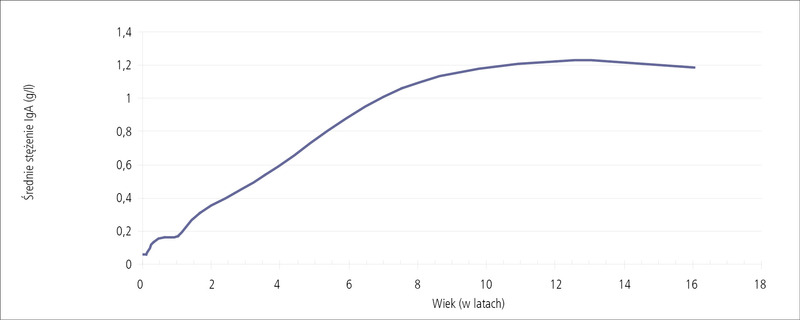
Rycina 3. Dojrzewanie syntezy przeciwciał IgA.
• Wywiad rodzinny wskazujący na zgony z powodu zakażeń we wczesnym dzieciństwie.
Ocena stężenia immunoglobulin
Badanie stężenia głównych klas immunoglobulin powinno być wykonane w pierwszej kolejności, co pozwoli na potwierdzenie lub wykluczenie najczęstszych niedoborów odporności. Ważne, aby wartości immunoglobulin porównać z referencyjnymi wartościami stężeń odpowiednimi dla wieku dziecka, które różnią się zasadniczo od stężeń immunoglobulin u dorosłych (tab. 4).7 Mniejsze stężenia immunoglobulin związane są z fizjologicznym, powolnym rozwojem układu odporności. Dotyczy to szczególnie immunoglobuliny IgA (ryc. 3). Dopiero po 4 roku życia u dziecka można rozpoznać ten niedobór, gdy stężenie IgA wynosi <0,07 g/l. Zdarzające się u tych dzieci zakażenia dróg oddechowych są na ogół spowodowane niedoborem podklas IgG.U dziecka w wieku powyżej 4 roku życia z niedoborem IgA w okresie rozwoju, a także w wieku dorosłym, mimo braku objawów klinicznych należy brać pod uwagę możliwość wystąpienia celiakii lub pospolitego zmiennego niedoboru odporności.
Cechy dysmorficzne w pierwotnych niedoborach odporności
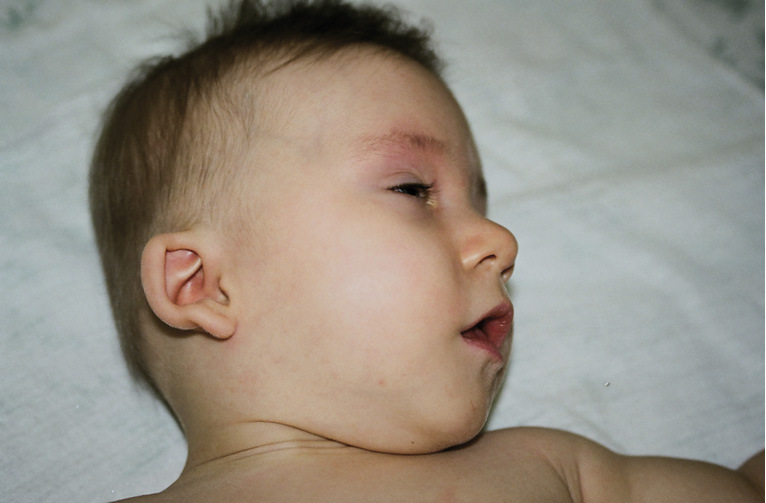
Rycina 4. Zespół DiGeorge’a.
W diagnostyce PNO ważne jest szczegółowe badanie kliniczne. Niektóre z PNO charakteryzują się występowaniem pewnych cech dysmorficznych, które mogą być pomocne w ustaleniu prawidłowego rozpoznania czy ukierunkowaniu dalszej diagnostyki.
Charakterystyczne cechy dysmorficzne w budowie twarzy obserwujemy głównie w zespołach: DiGeorge’a – zespół mikrodelecji 22q11.2, Nijmegen, hiper IgE – postać autosomalna dominująca, Blooma czy NEMO – jednej z postaci dysplazji ektodermalnej. PNO, jak już wspomniano, występują niezwykle rzadko, ale spotyka się je w praktyce klinicznej i należy o nich pamiętać.
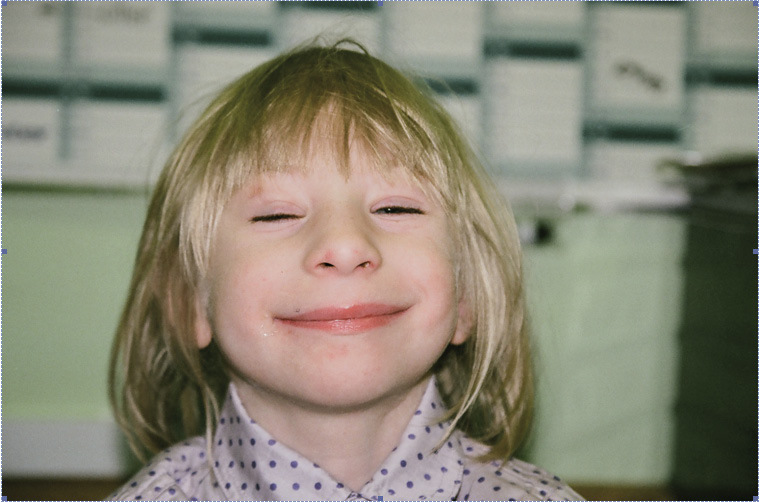
Rycina 5. Zespół Nijmegen.
Z wymienionych powyżej schorzeń najczęściej występuje zespół DiGeorge’a, spowodowany mikrodelecją prążka chromosomu 22q11. Częstość jego występowania ocenia się na 1/2000-1/4000 żywych urodzeń. W zespole DiGeorge’a dochodzi do zaburzeń rozwoju grasicy, jej aplazji lub hipoplazji, zwykle towarzyszy temu hipokalcemia wtórna do aplazji przytarczyc. Często obserwuje się wady serca, zaburzenia w rozwoju podniebienia i dysmorfię twarzy (ryc. 4). Cechy dysmorfii twarzy u dzieci z zespołem mikrodelecji 22q11 czasem mogą być subtelne, najczęściej obejmują: małe, nisko osadzone uszy, małe usta i cienkie wargi, wąskie szpary powiekowe, hiperteloryzm. Występuje też zmarszczka nakątna, podłużna twarz, mała cofnięta ku tyłowi żuchwa, malformacje w budowie i drożności nosa. Nieprawidłowości podniebienia obejmują: rozszczep wargi i/lub podniebienia, rozszczepiony albo podwójny języczek, rozszczep podśluzówkowy podniebienia, wysokie, gotyckie podniebienie, bardzo rzadko – rozszczep całkowity podniebienia (tzw. wilcza paszcza).8
Zespół Nijmegen, jeden z PNO przebiegających z zaburzeniami naprawy DNA, spowodowany jest mutacją w genie NBS1 znajdującym się na chromosomie 8q22, kodującym białko jądrowe – nibrynę. Cechami charakterystycznymi zespołu są: małogłowie, zahamowanie rozwoju fizycznego, niedobory odporności i znacznie zwiększone ryzyko wystąpienia nowotworu, głównie układu chłonnego. W rejestrze Europejskiego Towarzystwa Niedoborów Odporności znajduje się 149 chorych, większość z nich pochodzi z Europy Środkowej, głównie z Polski. Wiodącym objawem u dzieci z zespołem Nijmegen jest znaczne małogłowie, z obwodem głowy poniżej 3 centyla. Choroba objawia się zahamowaniem rozwoju wewnątrzmacicznego, a następnie niskorosłością. Cechy dysmorficzne twarzy obejmują: mikrognację, pochylone czoło, zadarty nos i dysmorficzne uszy (ryc. 5). Na skórze występują plamy „cafe-au-lait” oraz postępujące z wiekiem bielactwo.9,10
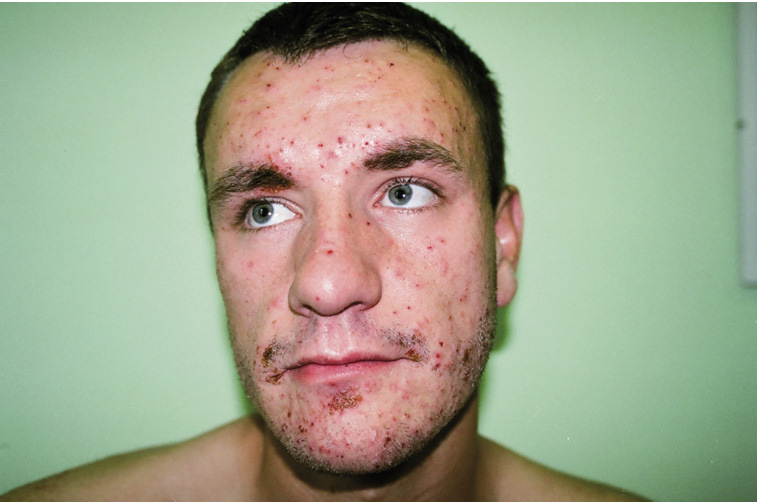
Rycina 6. Zespół hiper-IgE.
Zespół hiper IgE – postać autosomalna dominująca (AD-HIES) – jest bardzo rzadkim schorzeniem, występującym z częstością 1/500 000-1/1 000 000 żywych urodzeń. AD-HIES spowodowany jest mutacją w genie STAT3 znajdującym się na chromosomie 4q21.
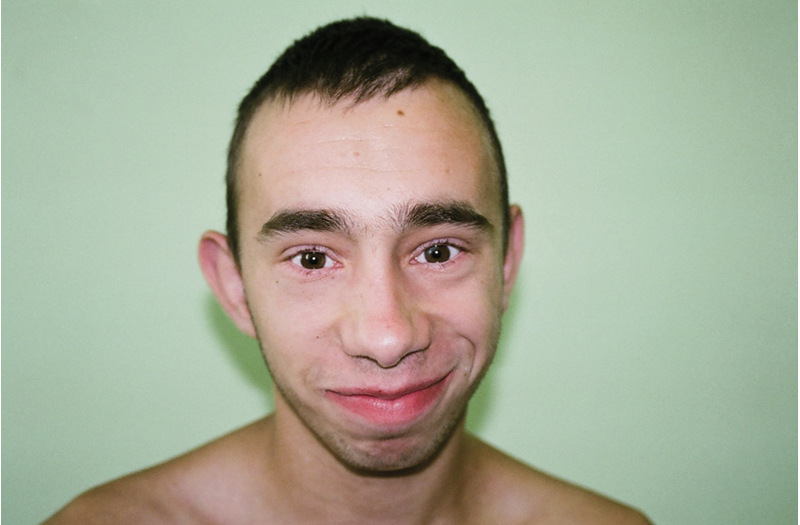
Rycina 7. Zespół Blooma.
W klasycznej postaci AD-HIES charakteryzuje się triadą objawów: nawracającymi gronkowcowymi ropniami skóry, zapaleniami płuc powikłanymi tworzeniem torbieli płucnych (pneumatocele), wypryskiem podobnym do atopowego oraz dużym stężeniem IgE (>2000 j.m./ml). Zmiany w układzie kostnym obejmują: patologiczne złamania, nadmierny przeprost w stawach, skoliozę, osteoporozę i przetrwałe zęby mleczne. Charakterystyczne zmiany dysmorficzne w obrębie głowy i twarzy to: grube rysy, głęboko osadzone oczy, wystające czoło, prognatyzm, pogrubiała dolna warga i małżowiny uszne, szeroka nasada nosa i gotyckie podniebienie (ryc. 6).11,12
Zespół Blooma jest wyjątkowo rzadkim PNO. Występuje najczęściej wśród Żydów aszkenazyjskich, charakteryzuje się rumieniem na twarzy (nasilającym się po ekspozycji na światło słoneczne), niedoborem wzrostu (już w okresie prenatalnym) oraz zwiększonym ryzykiem wystąpienia guzów litych i białaczek. Przyczyną zespołu jest mutacja w genie BLN, znajdującym się na chromosomie 15q26.1, kodującym białko należące do rodziny helikaz DNA. Główne objawy zespołu, poza niskim wzrostem i rumieniem na twarzy, to: długie nieproporcjonalne kończyny, przebarwienia na skórze typu plam „cafe-au-lait” i dysmorfia twarzy (ryc. 7). Twarz jest pociągła, wąska, z wydatnym nosem, obserwujemy hipoplazję policzków i niedorozwój żuchwy oraz duże odstające uszy, często wysoki ton głosu.
Erytrodermia – czy może wskazywać na pierwotny niedobór odporności?
U chorych z PNO mogą występować różnorodne zmiany skórne o charakterze przewlekłym i nawrotowym, często oporne na leczenie, m.in. owrzodzenia wokół odbytu i w jamie ustnej, ropnie skóry, pleśniawica czy zapalenie przyzębia. Wśród nich bardzo ważne miejsce zajmuje uogólniona złuszczająca erytrodermia, która może pojawić się zarówno już w pierwszych dniach życia, jak i na każdym etapie dalszego rozwoju dziecka. Dlatego też przyjmując w gabinecie niemowlę z erytrodermią, zawsze powinniśmy pomyśleć o wykluczeniu pierwotnego niedoboru odporności.
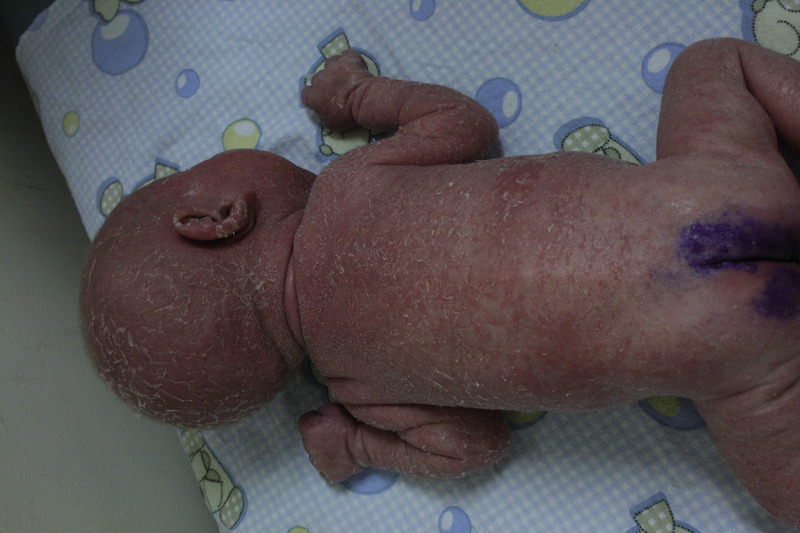
Rycina 8. Erytrodermia w atopowym zapaleniu skóry.