Przyczyny obwodowego PD zestawiono w tabeli 3.
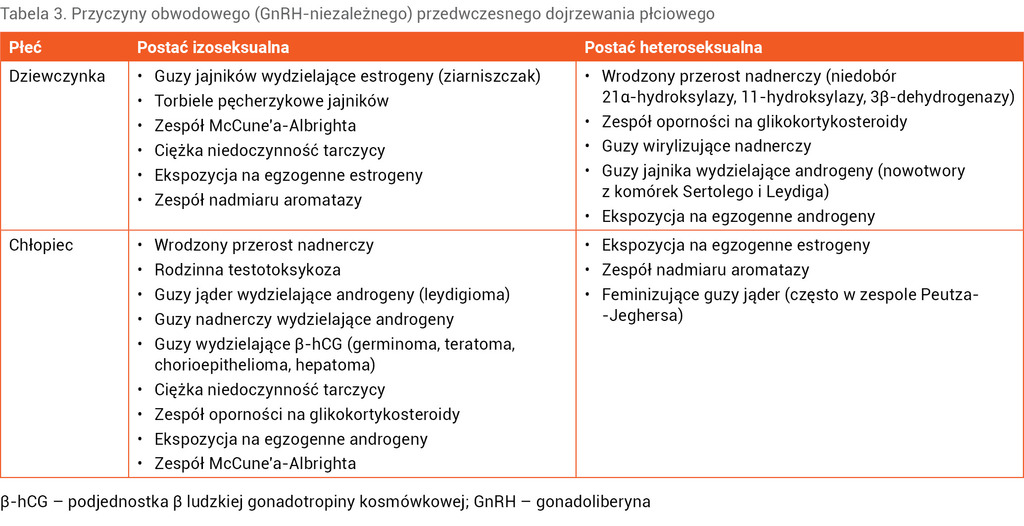
Tabela 3. Przyczyny obwodowego (GnRH-niezależnego) przedwczesnego dojrzewania płciowego
Wrodzony przerost nadnerczy
Najczęstszą przyczyną wrodzonego przerostu nadnerczy jest niedobór 21α-hydroksylazy powodujący zaburzenia steroidogenezy nadnerczowej. Zmniejszenie aktywności tego enzymu skutkuje hiperandrogenizmem, deficytem kortyzolu, a w cięższych postaciach klinicznych także niedoborem aldosteronu. Ze względu na obraz kliniczny, który jest zależny od stopnia niedoboru enzymu, WPN dzieli się na:
- postać klasyczną z utratą soli – najcięższy wariant choroby, w którym poza deficytem kortyzolu występuje również niedobór aldosteronu
- postać klasyczną bez utraty soli z wirylizacją prostą – w tej postaci aktywność enzymu wynosi 1-2% i obserwuje się zmniejszenie stężenia kortyzolu niewystarczające do zahamowania wydzielania ACTH, a czasami również nieznacznie niższe wartości aldosteronu. U dziewcząt objawy narastającej androgenizacji mogą zarówno występować po urodzeniu jako maskulinizacja zewnętrznych narządów płciowych, jak i ujawniać się w późniejszym wieku. U chłopców objawy izoseksualnego GnRH-niezależnego dojrzewania pojawiają się zazwyczaj między 3 a 7 rokiem życia. U dzieci tych obserwuje się znaczne przyspieszenie tempa wzrastania i akcelerację wieku kostnego, które w konsekwencji mogą prowadzić do niskiego wzrostu ostatecznego
- postać nieklasyczną (o późnym początku) – aktywność enzymu wynosi 50-70%, z reguły objawia się PD rozpoczynającym się po 5 roku życia od wystąpienia owłosienia płciowego i trądziku. Objawom mogą towarzyszyć niewielkie przyspieszenie tempa wzrastania i akceleracja wieku kostnego. U dziewcząt dodatkowo obserwuje się powiększenie łechtaczki, a u chłopców powiększenie prącia i ginekomastię. Wzrost ostateczny dziecka jest nieznacznie niższy od wzrostu rodziców. W badaniach hormonalnych stwierdza się podwyższone stężenia poranne 17-OHP (>10 ng/ml), testosteronu i androstendionu przy prawidłowych stężeniach ACTH i DHEAS. Stężenia gonadotropin są niskie. W przypadkach wątpliwych, gdy stężenie porannego 17-OHP wynosi 2-10 ng/ml, wskazane są wykonanie testu z analogiem ACTH i ocena przyrostu stężenia 17-OHP po stymulacji lub ocena profilu steroidowego w dobowej zbiórce moczu12,13. Leczenie WPN o późnym początku polega na substytucji hydrokortyzonu w celu obniżenia stężeń androgenów. U części pacjentów, szczególnie u chłopców, terapię można odstawić po osiągnięciu wzrostu ostatecznego. U dziewcząt i kobiet często leczenie wymaga kontynuacji ze względu na zaburzenia miesiączkowania, trądzik i hirsutyzm związane z nadmiarem androgenów.
Wprowadzone w 2016 roku badania przesiewowe u noworodków w kierunku klasycznej postaci niedoboru 21α-hydroksylazy znacznie przyczyniły się do wczesnego wykrywania choroby (przed wystąpieniem objawów niewydolności nadnerczy) oraz wdrożenia leczenia substytucyjnego. PD u dzieci z klasyczną postacią WPN jest oznaką niewystarczającego wyrównania hormonalnego (np. niesystematycznego leczenia, stosowania zbyt małej dawki hydrokortyzonu).
Zespół McCune’a-Albrighta
To zespół genetyczny, u podłoża którego leży somatyczna aktywująca mutacja genu GNAS1 kodującego powstawanie białka Gsα. Konsekwencją tej mutacji jest wzrost cyklicznego adenozynomonofosforanu (cAMP) w komórce, co prowadzi do proliferacji komórek i ich wzmożonej funkcji. Ponieważ do mutacji dochodzi po podziale zygoty, organizm pod względem mutacji jest mozaiką. Przekłada się to na różnorodny obraz kliniczny zespołu.
PD w przebiegu zespołu McCune’a-Albrighta występuje częściej u dziewcząt, a główną manifestacją endokrynologiczną są przedwczesne krwawienia z dróg rodnych, którym może towarzyszyć rozwój gruczołów piersiowych1-3. Krwawienia te stanowią konsekwencję tworzących się w jajnikach torbieli, które są źródłem estrogenów. W badaniach hormonalnych stwierdza się niskie podstawowe i stymulowane stężenia gonadotropin, przy wysokim stężeniu estradiolu. W obrazie USG miednicy mniejszej uwagę zwraca jednostronna lokalizacja zmian, co jest przyczyną dysproporcji w wielkości obu gonad2,10. Macica może być powiększona, z widocznym pogrubieniem endometrium. Objawy mają zazwyczaj cykliczny charakter i mogą prowadzić do przyspieszenia wieku kostnego. Dlatego też u dziewczynki z powtarzającymi się epizodami powiększenia piersi czy krwawieniami z dróg rodnych należy pomyśleć o diagnostyce w kierunku tego zespołu.
U chłopców PD występuje rzadziej (15%) i ma postać izolowanego powiększenia jąder, rzadziej owłosienia płciowego.
Typowymi składowymi zespołu poza PD są często jednostronne zmiany skórne o charakterze plam café au lait oraz dysplazja włóknista kości dotycząca głównie kości twarzoczaszki. Diagnoza jest zazwyczaj kliniczna, stawiana na podstawie obecności 2 z 3 powyższych objawów. Zespół kojarzy się również z wieloma innymi zaburzeniami dotyczącymi układu endokrynnego (takimi jak: nadczynność tarczycy, zespół Cushinga, gigantyzm), co należy uwzględniać w ocenie pacjenta.
W leczeniu PD w przebiegu zespołu McCune’a-Albrighta u dziewcząt stosuje się: leki antyestrogenowe (tamoksyfen), inhibitory aromatazy (letrozol) lub octan medroksyprogesteronu. Nie zaleca się chirurgicznego usuwania torbieli, gdyż w tym zespole mają one tendencję do nawracania.
Rodzinna testotoksykoza
To schorzenie spowodowane aktywującą mutacją genu dla receptora LH, która występuje zazwyczaj rodzinnie. Prowadzi ona do wydzielania testosteronu przez komórki Leydiga10,14. Choroba jest dziedziczona w sposób autosomalny dominujący. Manifestację kliniczną stwierdza się jedynie u chłopców, a objawy PD (owłosienie płciowe, powiększenie prącia, nieadekwatnie mały w stosunku do pozostałych cech płciowych symetryczny wzrost objętości jąder) pojawiają się u nich między 2 a 4 rokiem życia10,14. Ponadto obserwuje się przyspieszenie tempa wzrastania z niskim wzrostem ostatecznym. W leczeniu stosuje się leki antyandrogenowe w połączeniu z inhibitorami aromatazy10,14,15.
Zmiany nowotworowe
Pewne znaczenie w etiologii GnRH-niezależnego PD mają zmiany nowotworowe. Charakterystyczną cechą takiego PD jest duża dynamika narastania objawów oraz obecność znacznie podwyższonych stężeń hormonów: DHEAS w guzach nadnerczy i testosteronu w guzach jąder. U pacjentów z hormonalnie czynnymi nowotworami nadnerczy oprócz androgenizacji obserwuje się objawy zespołu Cushinga spowodowane wydzielaniem glikokortykosteroidów przez komórki guza. W nowotworach wywodzących się z komórek Leydiga cechom przedwczesnego pokwitania spowodowanego wysokim stężeniem testosteronu towarzyszy asymetria jąder. Ponadto GnRH-niezależne PD może wystąpić u chłopca z guzem germinalnym wydzielającym gonadotropinę kosmówkową (hCG – human chorionic gonadotropin). Na skutek reakcji krzyżowej cząsteczka hCG wiąże się z receptorem dla LH i pobudza komórki Leydiga do wydzielania testosteronu. U dziewcząt guzy te mogą pozostać długo nierozpoznane, ponieważ nie obserwuje się u nich klinicznych cech PD, gdyż do wydzielania estrogenów przez jajniki, oprócz obecności LH, konieczna jest stymulacja FSH16.
Niedoczynność tarczycy
Warto wspomnieć, że cechy PD mogą towarzyszyć niedoczynności tarczycy wskutek podobieństwa cząsteczki TSH do cząsteczek LH i FSH10. W długo trwającej ciężkiej niedoczynności tarczycy wysokie stężenie tyreoliberyny (TRH – thyrotropin-releasing hormone) może pobudzać wydzielanie TSH oraz FSH, a dodatkowo podwyższone stężenie TSH stymuluje receptory dla gonadotropin, co skutkuje powiększeniem gruczołów piersiowych u dziewcząt i jąder u chłopców. W tym typie dojrzewania nie obserwuje się przyspieszenia wzrastania, a wiek kostny jest opóźniony. Objawy PD ustępują po wyrównaniu niedoczynności tarczycy.