



Spis treści
- Wstęp
- Farmakologiczne mechanizmy działania analgetyków opioidowych
- Słabe opioidy
- Silne opioidy
- Opioidowe leki przeciwbólowe u ciężarnej (kategoria C wg FDA)
- Bezpieczeństwo stosowania analgetyków opioidowych
- Antagoniści receptorów opioidowych
Prawidłowo dobrane i dawkowane leki przeciwbólowe pozwalają na szybszą mobilizację pacjenta, zmniejszają częstość powikłań oraz prawdopodobieństwo przetrwałego bólu pozabiegowego.
Wstęp
Opioidy są jedną z głównych grup leków stosowanych w terapii bólu. Jak pokazuje doświadczenie, racjonalna farmakoterapia bólu, oparta na dobrej znajomości mechanizmów działania leków, ich farmakokinetyki, działań niepożądanych oraz interakcji z lekami z innych grup pozwala skutecznie łagodzić ból u większości,
bo aż u 85-95% pacjentów.
Farmakologiczne mechanizmy działania analgetyków opioidowych
Opioidy działają bezpośrednio na trzy typy receptorów opioidowych: μ (MOR), δ (DOR) oraz κ (KOR). Receptory opioidowe zlokalizowane są w strukturach ośrodkowego i obwodowego układu nerwowego. Należą one do rodziny receptorów sprzężonych z białkami G. W następstwie aktywacji, po przyłączeniu ligandu, receptor opioidowy zmienia swoją konformację, efektem czego jest przyłączenie swoistego białka błonowego związanego z GTP – białka G. Białko G składa się z trzech podjednostek: α, β i γ. Połączenie się białka G z odpowiednią domeną receptora opioidowego powoduje jego fosforylację i rozbicie na podjednostki. Podjednostka α białka G inicjuje dalsze skomplikowane procesy wewnątrzkomórkowe. W przypadku opioidu jego oddziaływanie z receptorem zlokalizowanym w błonie komórkowej powoduje zmianę właściwości związanego z nim białka Gi (pierwszy układ sygnałów) i zahamowanie aktywności cyklazy adenylanowej – enzymu odpowiedzialnego za produkcję cAMP w komórce (drugi układ sygnałów). Powoduje to zamknięcie kanałów wapniowych oraz aktywację transportu jonów potasu do przestrzeni zewnątrzkomórkowej, a w efekcie hiperpolaryzację błony komórkowej. W następstwie tych zmian zostaje wstrzymane uwalnianie pronocyceptywnych neuroprzekaźników oraz zwolnione lub zahamowane przewodzenie impulsów we włóknach nerwowych.
Opioidy stanowią grupę leków, które różnią się powinowactwem do trzech typów receptorów opioidowych (μ, δ, κ), rodzajem interakcji z receptorami opioidowymi (agoniści, częściowi agoniści, antagoniści), właściwościami fizykochemicznymi cząsteczki (wielkość, lipofilność), okresem półtrwania i charakterystyką farmakokinetyczną, a więc przebiegiem procesów wchłaniania, dystrybucji, metabolizmu i wydalania oraz oddziaływaniem na zstępujące układy kontroli bólu (tramadol, tapentadol) i receptory N-metylo-D-asparaginianowe (np. metadon).
Ze względu na siłę działania przeciwbólowego opioidy dzielimy na słabe i silne. W praktyce oznacza to, że słabe opioidy, klasyfikowane na drugim stopniu drabiny analgetycznej, charakteryzują się efektem pułapowym (tzn. powyżej pewnej dawki nie obserwujemy nasilenia działania analgetycznego, a zwiększa się ryzyko działań niepożądanych). Podział ten wydaje się szczególnie użyteczny w codziennej praktyce klinicznej. Dostępnymi w Polsce słabymi lekami opioidowymi są: kodeina, dihydrokodeina i tramadol, a do silnych opioidów stosowanych w naszym kraju należą: morfina, fentanyl, buprenorfina, metadon, tapentadol, oksykodon oraz połączenie oksykodonu z naloksonem.
Parametry farmakokinetyczne analgetyków opioidowych opisano w tabeli 1.
Słabe opioidy
Kodeina
Kodeina jest alkaloidem fenantrenowym, pochodną opium (3-metylomorfina), agonistą receptora opioidowego μ o sile działania 10-krotnie słabszej od morfiny. Jest metabolizowana przy udziale izoenzymu CYP2D6 na drodze O-demetylacji do morfiny oraz na drodze N-demetylacji do norkodeiny, która nie ma istotnego znaczenia klinicznego. Pierwszy szlak metaboliczny i powstająca w jego efekcie morfina odgrywa zasadniczą rolę w działaniu przeciwbólowym kodeiny. Jak pokazują badania, aktywność izoenzymu CYP2D6 jest bardzo zróżnicowana w zależności od populacji. Izoenzymu tego nie ma 6-14% osób rasy białej, 1-2% populacji azjatyckiej oraz 1-18% mieszkańców Afryki w zależności od rejonu, co czyni ich niewrażliwymi na kodeinę (tzw. wolny metabolizm). U kilku procent populacji obserwuje się natomiast jego nadekspresję (tzw. bardzo szybki metabolizm leku), w następstwie czego zachodzi szybsza przemiana kodeiny do morfiny. Chociaż wydaje się, że z uwagi na małe ilości morfiny powstającej z kodeiny ryzyko groźnych powikłań w tej grupie chorych jest niewielkie, nie wolno lekceważyć tego zjawiska.
Kodeina najczęściej jest podawana drogą doustną w postaci kropli lub tabletek o natychmiastowym uwalnianiu. Zaczyna działać po upływie 15-30 minut, a czas jej działania wynosi około 4 godzin. Zazwyczaj jest stosowana w dawce początkowej 20 mg co 4 godziny, maksymalna dawka dobowa wynosi 240 mg. Z uwagi na profil farmakokinetyczno-farmakodynamiczny (trudny do przewidzenia początek działania leku, długi czas latencji efektu analgetycznego) kodeina nie jest dzisiaj zalecana w leczeniu bólu. Wchodzi też w interakcje z wieloma lekami. Jest bezwzględnie przeciwwskazana przed ukończeniem 12 r.ż. z uwagi na ryzyko zgonu.
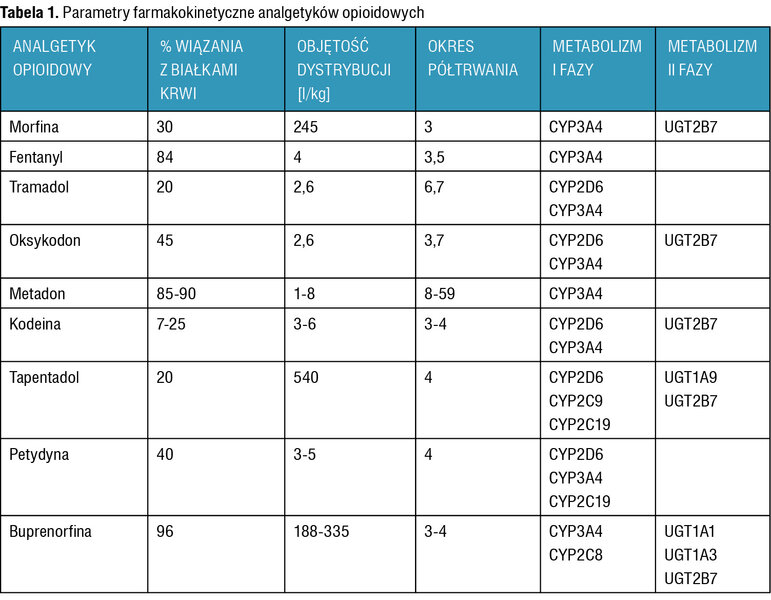
Tabela 1. Parametry farmakokinetyczne analgetyków opioidowych
Dihydrokodeina (DHC)
Dihydrokodeina jest pochodną kodeiny, która powstaje poprzez uwodornienie podwójnego wiązania w głównym łańcuchu cząsteczki kodeiny. Metabolizm leku jest podobny do metabolizmu kodeiny, ale w odróżnieniu od niej dihyrokodeina jest lekiem aktywnym. Działa z równą siłą także u pacjentów z uwarunkowanym genetycznie wolnym metabolizmem leków. Maksymalne stężenie w surowicy osiąga po około 3 godzinach od podania, a jej średni okres półtrwania wynosi 3,3-4,5 godziny. Dihydrokodeina jest dostępna w postaci tabletek o kontrolowanym uwalnianiu do stosowania co 12 godzin w dawkach 60 i 90 mg. Maksymalna dawka dobowa wynosi 240 mg.
Tramadol
Mechanizm działania
Jest syntetycznym lekiem przeciwbólowym o unikalnym mechanizmie działania. Tramadol jest czystym nieselektywnym słabym agonistą receptorów opioidowych μ, δ i κ, przy czym ma on szczególne powinowactwo do receptora μ. Inne mechanizmy działania analgetycznego wynikają z hamowania neuronalnego wychwytu zwrotnego noradrenaliny oraz zwiększenia uwalniania serotoniny. Siła działania tramadolu określana jest jako 1/10-1/6 siły działania morfiny.
Tramadol jest racemiczną mieszaniną dwóch enancjomerów. Enancjomery (-) oraz (+) wykazują działanie w układzie monoamin, a działanie na receptory opioidowe wykazuje jedynie enancjomer (+) O-demetylotramadolu, który słabiej niż np. tapentadol penetruje do OUN. Tym samym relatywny udział poszczególnych mechanizmów działania w efektach analgetycznych tramadolu może się zmieniać w czasie. Cząsteczka macierzysta ulega metabolizowaniu, tym samym mechanizm monoaminowy może się zmniejszać, a mechanizm opioidowy nasilać.
Po podaniu doustnym wchłania się ponad 90% tramadolu, średnia biodostępność wynosi 70% niezależnie od posiłku. Lek wiąże się z białkami w 20%. Okres półtrwania wynosi około 6 godzin niezależnie od drogi podania. Krzywa zależności dawka-efekt w zakresie dawek terapeutycznych ma przebieg liniowy.
W badaniach eksperymentalnych tramadol, ale nie jego główny metabolit, wykazywał działanie hamujące potencjał czynnościowy w izolowanych nerwach. Jest to działanie podobne do leków znieczulenia miejscowego i w przypadku tramadolu nie było zależne od działania na receptor opioidowy ani na układ monoamin.
Tramadol jest lekiem przeciwbólowym o unikalnym, podwójnym mechanizmie działania. Analgezja po podaniu tramadolu wynika zarówno z oddziaływania tego leku na receptory opioidowe μ w OUN, jak i z aktywacji zstępujących układów antynocyceptywnych: noradrenergicznego i serotoninergicznego. Za efekt opioidowy leku odpowiada O-demetylotramadol – metabolit powstający przy udziale CYP2D6, mający 700 razy większe powinowactwo do receptora opioidowego μ i siłę działania kilkakrotnie przewyższającą związek macierzysty. Drugim metabolitem tramadolu jest nieaktywny N-metylotramadol, powstający przy udziale CYP3A4. 90% tramadolu i jego metabolitów jest wydalane przez nerki. Okres półtrwania tramadolu wynosi 6 godzin, jego aktywnego metabolitu 7,5 godziny. W niewydolności wątroby i nerek okres półtrwania tramadolu i jego aktywnej pochodnej wydłuża się 2-3-krotnie. Z tego względu u chorych z ciężką niewydolnością wątroby i nerek zaleca się zmniejszenie dawek oraz wydłużenie odstępów między kolejnymi dawkami leku. Mniejszą skuteczność tramadolu, podobnie jak w przypadku kodeiny, obserwuje się w populacji osób wolno metabolizujących lek, chorych otrzymujących równocześnie inhibitory CYP2D6 (np. duloksetynę, fluoksetynę, paroksetynę, sertralinę, cytalopram, haloperydol), ondansetron oraz karbamazepinę. Także w przypadku nudności i wymiotów u pacjentów stosujących tramadol przeciwwskazane jest stosowanie metoklopramidu, który hamując aktywność CYP2D6, utrudnia metabolizm tramadolu do bardziej aktywnego metabolitu i przedłuża równocześnie czas działania leku macierzystego. Osłabienie efektu analgetycznego tramadolu występuje w przypadku równoczesnego stosowania ondansetronu, a także mirtazapiny, co tłumaczy się przede wszystkim antagonizmem tych leków w stosunku do receptorów serotoninowych, natomiast w przypadku równoczesnego podawania karbamazepiny, która jest silnym induktorem CYP3A4 – nasilenie przemiany tramadolu do nieaktywnego metabolitu N-demetylotramadolu.
Wskazania
Tramadol wykazywał skuteczność analgetyczną zarówno w ostrym bólu pooperacyjnym i pourazowym, jak i przewlekłym w badaniach eksperymentalnych, a także w badaniach klinicznych. Jest skuteczny w leczeniu bólu neuropatycznego – zmniejsza nasilenie bólu, parestezje, allodynię i ból wzbudzany. Współczynnik NNT obliczony na podstawie dostępnych randomizowanych badań klinicznych określono na 3,8, a współczynnik NNH na 8,3.
Wskazaniem do zastosowania tramadolu jest leczenie bólu o nasileniu od umiarkowanego do silnego. U pacjentów w podeszłym wieku konieczność modyfikacji dawki zachodzi w przypadku niewydolności wątroby lub nerek, zaleca się wówczas wydłużenie odstępu między dawkami. Lek nie powinien być stosowany u pacjentów z ciężką niewydolnością nerek lub wątroby, a w przypadku niewydolności tych narządów w stopniu umiarkowanym zaleca się wydłużenie odstępu pomiędzy dawkami.
Interakcje
W badaniach klinicznych dotyczących ostrego bólu pooperacyjnego obliczono NNT dla dawek 50, 100 i 150 mg tramadolu – wynoszą one odpowiednio 7,1, 4,8 oraz 2,4. Ponadto w badaniach eksperymentalnych i klinicznych wykazano efekt synergistyczny przy łącznym stosowaniu w terapii ostrego bólu tramadolu z paracetamolem (ból niezapalny) oraz tramadolu z deksketoprofenem (ból zapalny).
Tramadol w skojarzeniu z takimi lekami, jak SSRI i inhibitory MAO, może spowodować groźny dla życia zespół serotoninowy. Ma to związek z oddziaływaniem leku na układ monoamin. Tramadol może nasilać również działanie TCA i leków przeciwpsychotycznych.
Należy zachować ostrożność podczas jednoczesnego podawania tramadolu i pochodnych kumaryny ze względu na ryzyko niekontrolowanego zwiększenia INR i powstania wybroczyn i krwawień.
Stosowanie przez kobiety w ciąży
Tramadol przenika przez łożysko, a jego bezpieczeństwo u kobiet ciężarnych nie zostało ustalone, dlatego nie zaleca się jego stosowania w czasie ciąży. Nie wpływa na czynność skurczową macicy. U noworodków matek długotrwale przyjmujących tramadol w ciąży może wystąpić zespół odstawienny. Tramadol przenika do mleka kobiecego, dlatego nie powinien być stosowany długotrwale w okresie laktacji. Jednorazowe podanie leku matce karmiącej nie ma znaczenia klinicznego i nie jest konieczne przerwanie karmienia piersią, aczkolwiek ostatnie badania sugerują odstąpienie od stosowania tramadolu przez kobiety karmiące.
Działania niepożądane
Najczęstszymi działaniami niepożądanymi, szczególnie w początkowej fazie terapii tramadolem, są nudności i wymioty (u 10% pacjentów) oraz nadmierne pocenie się. Ich nasilenie zazwyczaj zmniejsza się z upływem czasu. Z oddziaływaniem tramadolu na układ monoamin wiąże się ryzyko groźnego dla życia zespołu serotoninowego u chorych otrzymujących tramadol w skojarzeniu z innymi lekami hamującymi wychwyt zwrotny serotoniny, takimi jak sertralina, cytalopram, escytalopram, paroksetyna, wortioksetyna.
Tramadol w zalecanych dawkach może upośledzać zdolność prowadzenia pojazdów mechanicznych, szczególnie w połączeniu z alkoholem i innymi substancjami psychotropowymi.
Często (≥1/100, ≤1/10) występują także bóle głowy, senność, zaparcia, suchość jamy ustnej, nadmierna potliwość i zmęczenie. Objawy ze strony OUN mogą się nasilić w przypadku jednoczesnego stosowania innych leków hamujących OUN lub alkoholu. Częstość zaparć i ryzyko uzależnienia podczas leczenia tramadolem są znacznie mniejsze niż w przypadku silnych opioidów.
W przypadku zatrucia należy pamiętać, że lek w niewielkim stopniu jest eliminowany przy zastosowaniu hemodializy czy hemofiltracji.
Tramadol należy podawać ostrożnie pacjentom uzależnionym od opioidów, po urazie głowy, we wstrząsie, z zaburzeniami świadomości niejasnego pochodzenia, zaburzeniami oddechowymi oraz w przypadku podwyższonego ciśnienia wewnątrzczaszkowego. Tramadol może obniżać próg drgawkowy, ryzyko drgawek zwiększa się, gdy pacjent stosuje dawkę >400 mg/24 h, a także przy równoczesnym stosowaniu leków obniżających próg drgawkowy. Cechuje go bardzo słaby potencjał lekozależności oraz rozwoju tolerancji, ale u pacjentów nadużywających leków tramadol powinien być stosowany krótkotrwale i pod ścisłym nadzorem lekarza.
Różnice w metabolizowaniu leku
Tramadol jest metabolizowany w procesie N- i O-demetylacji oraz sprzęgania produktów O-demetylacji z kwasem glukuronowym. Za efekt analgetyczny odpowiada O-demetylotramadol – główny metabolit tramadolu powstający przy udziale CYP2D6. Metabolit ten jest 2-4 razy silniejszy od substancji macierzystej. Izoenzym 2D6 jest polimorficzny, a jego genetycznie uwarunkowana odmiana może skutkować niepełną funkcją, którą obserwujemy u 6-14% populacji kaukaskiej, lub też nadmierną aktywnością. U osób słabo metabolizujących nie dochodzi do przekształcenia tramadolu w aktywny metabolit i tym samym skuteczność leku jest osłabiona, z kolei u osób bardzo szybko metabolizujących mogą pojawić się objawy toksyczne nawet po małej dawce opioidu. Osłabienie efektu analgetycznego tramadolu w przypadku równoczesnego podawania ondansetronu tłumaczy się przede wszystkim antagonizmem ondansetronu do receptorów serotoninowych 5-HT3, a w przypadku równoczesnego podawania karbamazepiny, która jest silnym induktorem CYP3A4, nasileniem przemiany tramadolu do nieaktywnego metabolitu N-demetylotramadolu. Według ChPL inne leki hamujące CYP3A4, np. ketokonazol i erytromycyna, mogą hamować metabolizm tramadolu przez N-demetylację i prawdopodobnie metabolizm aktywnego O-demetylotramadolu, ale znaczenie kliniczne tych interakcji nie zostało dokładnie wyjaśnione. Około 85% wchłoniętego leku jest metabolizowane w wątrobie i wydalane z moczem (90%) i kałem (10%). U pacjentów z niewydolnością nerek lub wątroby okres półtrwania tramadolu może ulec niewielkiemu wydłużeniu.
Postacie leku i dawkowanie
Tramadol jest opioidowym lekiem przeciwbólowym, który cechuje stosunkowo niski potencjał wywoływania lekozależności oraz rozwoju tolerancji. Preparaty doustne o natychmiastowym uwalnianiu (kapsułki, krople) stosuje się w dawce 25-50 mg (do 100 mg), co 4, 6, 8 godzin, a preparaty o przedłużonym działaniu w dawce 50-200 mg co 12 godzin. Dawka dobowa tramadolu nie powinna przekraczać 400 mg/24 h. W szczególnych sytuacjach klinicznych, np. u chorych na nowotwór, dawkę można zwiększyć do 600 mg/24 h. Zastosowanie preparatu w kroplach (20 kropli = 50 mg) pozwala na skuteczne postępowanie przeciwbólowe, nie zaleca się go natomiast z uwagi na profil farmakokinetyczny. Ta postać leku powinna być zarezerwowana dla pacjentów, którzy mają trudności w połykaniu. Tramadol jest dostępny również w połączeniu z paracetamolem oraz z deksketoprofenem. Takie preparaty przyspieszają działanie leku i wykazują synergistyczny efekt analgetyczny. Tramadol jest także dostępny w preparatach do podawania drogą dożylną. Terapię rozpoczyna się zazwyczaj od dawki 25-50 mg podawanej co 4, 6, 8 godzin. Dawka dobowa tramadolu nie powinna przekroczyć 400 mg/24 h. Tramadolu nie należy łączyć z cyproheptadyną (działanie antyserotoninowe) ani z mirtazapiną (antagonista receptora 5-HT3) z uwagi na antagonizowanie działania przeciwbólowego leku.
Silne opioidy
Morfina
Mechanizm działania
Morfina jest czystym agonistą receptora opioidowego μ. Następstwem tego jest liniowa zależność pomiędzy podaną dawką a efektem przeciwbólowym. Średni czas działania morfiny wynosi 4 godziny. Morfina podlega procesom metabolicznym w wątrobie, ścianie jelit, nerkach i OUN przede wszystkim na drodze sprzęgania z kwasem glukuronowym przy udziale glukuronylotransferazy. Jej głównymi metabolitami są morfino-3-glukuronian (M3G) i morfino-6-glukuronian (M6G). Niewielka część morfiny (<5%) jest metabolizowana przy udziale CYP450 do normorfiny. Morfino-6-glukuronian jest aktywnym metabolitem morfiny o powinowactwie do receptorów opioidowych μ i sile działania wielokrotnie przekraczającej siłę przeciwbólowego działania morfiny. Drugi z metabolitów, morfino-3-glukuronian, nie wykazuje działania przeciwbólowego, ma natomiast właściwości neurotoksyczne potwierdzone w badaniach na zwierzętach. Jego nagromadzenie może prowadzić do zaburzeń poznawczych, delirium, pobudzenia i mioklonii. Po podaniu doustnym z uwagi na efekt pierwszego przejścia stosunek stężenia M3G do M6G i morfiny jest znacznie wyższy niż u pacjentów otrzymujących morfinę parenteralnie. Ze względu na fakt, że w warunkach prawidłowych do 30% morfiny jest metabolizowane pozawątrobowo, umiarkowanego stopnia uszkodzenie wątroby nie zakłóca w sposób istotny procesów jej metabolizmu. U chorych z marskością wątroby obserwuje się zwiększenie biodostępności morfiny (wzrost jej stężenia w surowicy po podaniu określonej dawki), prawdopodobnie w następstwie zmniejszenia przepływu wątrobowego krwi. Z kolei u chorych z nieprawidłową czynnością nerek upośledzona jest nerkowa eliminacja metabolitów morfiny, a okres półtrwania morfino-6-glukuronianu trzykrotnie się wydłuża. Z tego względu chorzy z nieprawidłową czynnością nerek są bardziej narażeni na działania niepożądane morfiny i wymagają w związku z tym ścisłego monitorowania, redukcji stosowanych dawek, wydłużenia czasu między kolejnymi dawkami, zmiany drogi podawania z doustnej na podskórną lub najlepiej zamiany morfiny na inny opioid, którego metabolity wydalane są drogą przewodu pokarmowego (np. na buprenorfinę, metadon, fentanyl).
Morfina skutecznie uśmierza ból, zwłaszcza tępy, ale jest mniej skuteczna w bólu nagłym, przeszywającym, co powinno zostać uwzględnione przy wyborze leku. Wywołuje dobry nastrój, euforię (rzadziej dysforię), obniża zdolność koncentracji i zwalnia procesy myślowe. Działa depresyjnie na ośrodek oddechowy – zmniejsza częstość oddychania (do bezdechu włącznie, ale w dawkach ponadterapeutycznych lub w przypadku pogorszenia funkcji wydalniczej nerek), powoduje także depresję ośrodka kaszlu oraz pobudza jądra parasympatyczne nerwu okoruchowego, co powoduje zwężenie źrenic (źrenice szpilkowate). Pobudza także strefę chemoreceptorową pnia mózgu, czego następstwem są nudności i wymioty obserwowane szczególnie często w początkowym okresie terapii. Może uwalniać histaminę, czego efektem jest obserwowany czasami po jej zastosowaniu świąd skóry. Wzmaga napięcie mięśniówki przewodu pokarmowego, zwłaszcza zwieraczy, oraz osłabia perystaltykę jelit, co prowadzi do zaburzeń defekacji. Morfinę stosuje się zarówno drogą doustną, jak i parenteralnie, jednak wchłanianie leku z przewodu pokarmowego jest istotnie słabsze, dlatego aby uzyskać równoważny efekt przeciwbólowy, drogą doustną trzeba podawać 2-3-krotnie większe dawki niż parenteralnie. Przedawkowanie morfiny objawia się zaburzeniami świadomości, obniżeniem ciśnienia tętniczego krwi oraz zwolnieniem rytmu oddechowego do bezdechu włącznie. Ból jest naturalnym antagonistą tych objawów niepożądanych.
Interakcje
Łączne podawanie morfiny z benzodiazepinami lub innymi lekami o działaniu depresyjnym na OUN zwiększa ryzyko głębokiej sedacji, hipotonii, delirium, a także depresji ośrodka oddechowego. Wiele leków stosowanych równocześnie z morfiną nasila zaburzenia defekacji. Są to m.in. leki antycholinergiczne i antagoniści receptora 5-HT3. Ryzyko mioklonii zwiększa się z kolei podczas łącznego stosowania morfiny z fenotiazynami, NLPZ oraz lekami przeciwdepresyjnymi. Morfiny nie należy łączyć z diklofenakiem, aceklofenakiem oraz nilotynibem, gdyż leki te hamują jej glukuronidację i spowalniają eliminację z ustroju.
Sposób podania i dawkowanie
Morfina jest podawana doustnie w preparatach o natychmiastowym uwalnianiu (roztwór, tabletki) zazwyczaj w dawce początkowej 5-10 mg co 4 godziny lub w preparacie o zmodyfikowanym uwalnianiu w dawce początkowej 20-30 mg co 12 godzin. Jest także stosowana parenteralnie: podskórnie, dożylnie lub okołordzeniowo (zewnątrzoponowo, podpajęczynówkowo). Może być ponadto stosowana miejscowo, najlepiej w postaci żelu, m.in. na trudno gojące się rany, zmiany troficzne, czy w postaci płukanek u chorych z zapaleniem błony śluzowej jamy ustnej po naświetlaniach głowy i szyi. Jest lekiem z wyboru w leczeniu bólu z dusznością u chorych na nowotwór.
Fentanyl
Mechanizm działania
Fentanyl jest czystym agonistą receptora opioidowego μ, który jako pierwszy z opioidów został zastosowany w postaci przezskórnej do leczenia bólu przewlekłego. W Polsce został zarejestrowany w 1995 roku. Dzięki małej masie cząsteczkowej oraz dużej lipofilności fentanyl łatwo przechodzi przez skórę i z krwiobiegu szybko przedostaje się przez barierę krew-mózg do OUN, gdzie łączy się z receptorami opioidowymi μ (po 3-4 minutach jest za barierą krew-mózg). Jest on metabolizowany przy udziale cytochromu CYP3A4 głównie do nieaktywnego norfentanylu, a następnie wydalany z moczem w postaci nieaktywnych metabolitów i w 7-10% w postaci niezmienionej. CYP3A4 jest enzymem odpowiedzialnym za metabolizm ponad połowy wszystkich stosowanych leków, a wiele z nich poprzez hamowanie lub aktywację CYP3A4 może wpływać na metabolizm fentanylu i ostateczny efekt po jego zastosowaniu. Do inhibitorów CYP3A4 zaliczamy m.in. cyprofloksacynę, erytromycynę, klarytromycynę, flukonazol, ketokonazol, midazolam, omeprazol, fluoksetynę, paroksetynę. Łączne zastosowanie któregokolwiek z tych leków z fentanylem może prowadzić do wzrostu stężenia fentanylu we krwi i wystąpienia objawów niepożądanych, w tym ciężkich, do zgonu włącznie. Nie zaleca się także kojarzenia fentanylu z imatynibem oraz nilotynibem z uwagi na ryzyko niekorzystnych interakcji farmakokinetycznych prowadzących do zwiększenia toksyczności fentanylu. Z kolei dodanie do fentanylu leku indukującego CYP3A4, takiego jak karbamazepina, fenobarbital, deksametazon, fenytoina lub ryfampicyna, może być przyczyną gorszego efektu przeciwbólowego fentanylu w następstwie przyspieszenia jego przemiany do nieaktywnych metabolitów. Modyfikacja farmakokinetyki fentanylu przez inne, stosowane równolegle leki jest szczególnie ważna w przypadku stosowania preparatów przezśluzówkowych tego leku ze względu na szybki wzrost stężenia fentanylu we krwi, co przy braku adaptacji ośrodka oddechowego może być przyczyną zagrażających życiu powikłań (depresji oddechowej). Fentanyl jest dość dobrze tolerowany przez pacjentów z umiarkowaną niewydolnością wątroby i nerek. U chorych z wyrównaną niewydolnością wątroby i nerek nie stwierdza się istotnego wydłużenia okresu półtrwania leku, natomiast w ciężkiej niewydolności nerek parametr ten dwukrotnie się wydłuża. Należy podkreślić, że chorzy z upośledzoną czynnością wątroby i nerek leczeni fentanylem wymagają dokładnego nadzoru i monitorowania bezpieczeństwa terapii z uwagi na ryzyko stopniowej kumulacji leku. Fentanyl nie ma działania obwodowego odpowiedzialnego za niekorzystny wpływ zwiększający napięcie mięśni gładkich przewodu pokarmowego, czym tłumaczy się mniej nasilone zaburzenia defekacji i działanie prowymiotne. W porównaniu z morfiną fentanyl ma słabsze działanie sedacyjne i w niewielkim stopniu uwalnia histaminę. Uważa się, że stosowanie fentanylu w postaci przezskórnej zapewnia stały poziom leku w surowicy krwi, co pozwala na utrzymanie stabilnego poziomu analgezji, przyczynia się także do zmniejszenia częstości bólów przebijających (nawet o około 20%).
Sposób podania i dawkowanie
Na rynku dostępne są plastry uwalniające fentanyl w dawce 12,5, 25, 50, 75 i 100 μg/h, czas działania plastra wynosi 3 doby. Ze stosowaniem fentanylu związane jest ryzyko wystąpienia zespołu serotoninowego. Fentanyl w postaci tabletek podpoliczkowych, podjęzykowych oraz preparatu donosowego jest wykorzystywany w leczeniu bólów przebijających w przebiegu choroby nowotworowej. W preparacie w postaci tabletek podpoliczkowych zastosowano technologię umożliwiającą szybką absorpcję opioidu przez błonę śluzową. Dostępne są tabletki zawierające 100, 200, 400, 600 i 800 μg substancji czynnej. W przypadku preparatu donosowego 1 dawka leku zawiera 50 μg fentanylu. Początkowa dawka powinna wynosić jednorazowo 50 μg do jednego nozdrza, w razie konieczności dawkowanie można zwiększać. Jeżeli nie uzyskano efektu przeciwbólowego, lek można powtórnie zastosować najwcześniej po 10 minutach. Fentanylu nie należy stosować jednocześnie z nilotynibem i imatynibem z uwagi na działania niepożądane, jakie mogą wystąpić w konsekwencji ich interakcji.
Buprenorfina
Mechanizm działania
Buprenorfina jest syntetycznym lekiem opioidowym, pochodną tebainy. Jest agonistą receptora opioidowego μ i nocyceptynowego oraz antagonistą receptora opioidowego δ i κ. Z tego powodu nie zaleca się jej łączenia z oksykodonem. Buprenorfina jest częściowym agonistą receptora opioidowego μ. Cechą charakterystyczną częściowych agonistów receptorów opioidowych jest tzw. efekt pułapowy, co oznacza, że po przekroczeniu pewnej dawki działanie analgetyczne leku się zmniejsza. Jednakże w badaniach klinicznych nie zaobserwowano efektu pułapowego dla buprenorfiny, pomimo stosowania jej w dużych dawkach. W badaniach eksperymentalnych w dawkach <10 mg/kg m.c. krzywa zależności dawka-efekt jest linią prostą, tzn. każdemu zwiększeniu dawki buprenorfiny towarzyszy nasilenie efektu analgetycznego, co jest charakterystyczne dla czystego agonisty. W dawkach >10 mg/kg m.c. obserwowano zmniejszanie się efektu analgetycznego, ale jedynie w niektórych testach behawioralnych w badaniach eksperymentalnych. Dawka ta znacznie przekracza stosowaną w praktyce klinicznej. Wykazuje jednak efekt pułapowy w odniesieniu do depresyjnego działania na ośrodek oddechowy, w związku z czym prawdopodobieństwo depresji oddechowej po jej zastosowaniu jest niewielkie. Buprenorfina ma wysokie powinowactwo do receptora opioidowego μ, wyższe niż morfina, a także cechuje się dużą aktywnością wewnętrzną po połączeniu z receptorem. W badaniach eksperymentalnych efekt analgetyczny uzyskiwano przy zajęciu mniej niż 100% receptorów opioidowych, co pozwala na stosowanie w razie potrzeby innych opioidów. Efekt analgetyczny, jak również działania niepożądane są całkowicie odwracalne po podaniu naloksonu. W badaniach eksperymentalnych buprenorfina wykazuje działanie analgetyczne na poziomie rdzeniowym – działa na receptory opioidowe, podobnie jak morfina i fentanyl. Natomiast na poziomie ponadrdzeniowym działa w innym mechanizmie niż opioidowy i odmiennym niż morfina i fentanyl. Ten szczególny mechanizm działania na poziomie mózgowia może tłumaczyć mniejsze ryzyko depresji oddechowej w porównaniu z innymi opioidami, minimalny wpływ na funkcje poznawcze u osób starszych, a także małe ryzyko rozwoju tolerancji i uzależnienia. Małe ryzyko powstania tolerancji powoduje, że w przeciwieństwie do innych czystych agonistów podczas stosowania buprenorfiny znacznie wolniej można zwiększać dawkę leku.
Nocyceptyna i jej receptor odgrywają istotną rolę w procesie nocycepcji, rozwoju tolerancji i uzależnienia, dlatego unikalne agonistyczne działanie buprenorfiny w stosunku do receptora nocyceptynowego może mieć znaczenie w efektach klinicznych.
W badaniach eksperymentalnych i klinicznych buprenorfina silnie hamuje hiperalgezję, a co ciekawe, efekt ten występuje wcześniej niż efekt analgetyczny i trwa znacznie dłużej, nie udało się jednak w pełni wyjaśnić jego mechanizmu. W badaniach eksperymentalnych wykazano, że buprenorfina blokuje kanały sodowe regulowane napięciem.
Sposób podania i dawkowanie
Wskazaniem do stosowania buprenorfiny w postaci tabletek podjęzykowych po 0,2 i 0,4 mg oraz ampułek 0,3 mg jest ból różnego pochodzenia o średnim i dużym nasileniu. Natomiast w przypadku postaci przezskórnej (35, 52,5, 70 μg/h) jest to ból o średnim i dużym nasileniu w przebiegu chorób nowotworowych i ból o dużym nasileniu w przebiegu innych schorzeń, jeżeli nie ustępuje po zastosowaniu nieopioidowych leków przeciwbólowych. W Europie dostępna jest również buprenorfina w postaci przezskórnej w małych dawkach 5, 10 i 20 μg/h stosowanych co 7 dni.
Buprenorfina działa 75-115-krotnie silniej niż morfina. Przy określaniu dawek równoważnych należy brać pod uwagę wskazania kliniczne, sposób podania oraz zmienność osobniczą z uwagi na profil leku. Buprenorfina dobrze wchłania się z błony śluzowej jamy ustnej oraz z przewodu pokarmowego, gdzie jednak ulega inaktywacji w jelitach i wątrobie, dlatego ta droga podania nie jest zalecana. Może być podawana domięśniowo lub w powolnym wlewie dożylnym.
Po podaniu podjęzykowym buprenorfina szybko wchłania się przez śluzówkę jamy ustnej, a jej maksymalne stężenie w osoczu występuje po 90 min, co powoduje, że lek podany tą drogą nie powinien być stosowany w przypadku bólu ostrego ani bólu przebijającego u chorego na nowotwór. Eliminacja zachodzi wolno, do 20-25 godzin. Tabletek podjęzykowych nie należy rozgryzać ani połykać. Dawkowanie u dzieci po 12 r.ż. i dorosłych wynosi od 0,2 do 0,4 mg co 6-8 godzin. Dawkowanie u dzieci w wieku od 6 do 12 r.ż. zależy od masy ciała: 16-25 kg – 0,1 mg, >25-37,5 kg – 0,1-0,2 mg, >37,5-50 kg – 0,2-0,3 mg. Dawki te należy podawać co 6-8 godzin. Nie zaleca się podawania tabletek dzieciom przed ukończeniem 6 r.ż.
Przy przyklejeniu systemu przezskórnego buprenorfina jest wchłaniana do krążenia przez skórę, co następuje poprzez kontrolowane uwalnianie z matrycy adhezyjnej. Po 12-24 godzinach występuje minimalne stężenie skuteczne, czas działania to 60-80 godzin. Po usunięciu systemu przezskórnego stężenie buprenorfiny w osoczu zmniejsza się i ulega ona wydaleniu ze średnim okresem półtrwania około 30 godzin, wydalanie to jest wolniejsze niż po podaniu dożylnym. Nie przeprowadzono badań nad stosowaniem leku w postaci przezskórnej u osób <18 r.ż.
Dawkowanie buprenorfiny w postaci przezskórnej u osób po 18 r.ż. zależy od osobniczej wrażliwości pacjenta. U pacjentów, u których nie stosowano dotychczas żadnych analgetyków lub tylko analgetyki nieopioidowe, leczenie należy rozpocząć od plastra w najmniejszej dawce 35 μg/h. W przypadku pacjentów, u których stosowano już silne opioidy, należy dobrać dawkę plastra w odniesieniu do dawki dobowej i rodzaju uprzednio stosowanego opioidu, pomocne są tabele dawek równoważnych. Leczenie pacjentów przyjmujących uprzednio dawki silnych opioidów odpowiadające 120 mg/24 h morfiny p.o. można rozpocząć od 52,5 μg/h. Istnieją pojedyncze doniesienia opisujące stosowanie plastra w dawce 210 μg/h, podczas którego nie zaobserwowano efektu pułapowego dla analgezji.
Po zastosowaniu po raz pierwszy leku w postaci przezskórnej stężenie buprenorfiny w surowicy zwiększa się powoli, pierwszej oceny działania należy dokonać po upływie 24 godzin od przyklejenia plastra. Uprzednie leczenie przeciwbólowe, z wyjątkiem opioidowych systemów przezskórnych, należy kontynuować podczas pierwszych 12 godzin po naklejeniu plastra, a w ciągu następnych 12 godzin można podać krótko działający lek przeciwbólowy. Pojedynczy system przezskórny należy zmieniać co 96 godzin, a każdy następny umieszczać w innym miejscu na skórze. Jednocześnie można używać maksymalnie dwu plastrów niezależnie od dawki. Pacjenci wymagający dodatkowych leków przeciwbólowych do opanowania bólu mogą stosować tabletki podjęzykowe buprenorfiny w dawce 0,2-0,4 mg. Plastry należy umieszczać na płaskich, nieowłosionych obszarach skóry górnej połowy ciała. Przed przyklejeniem plastra owłosienie należy usunąć nożyczkami, w razie potrzeby skórę umyć wodą. W trakcie noszenia plastra można się kąpać, brać prysznic i pływać, nie należy poddawać go działaniu gorąca. Po usunięciu plastra stężenie buprenorfiny zmniejsza się powoli, nie ma jednak informacji dotyczących początkowej dawki innych opioidów po odstawieniu plastra.
Buprenorfinę w ampułkach można podawać domięśniowo lub w powolnym wstrzyknięciu dożylnym. W bólach różnego pochodzenia dawka dla dorosłych i dzieci >12 lat wynosi zazwyczaj 0,3-0,6 mg co 6-8 h; dzieci <12 lat: 3-6 µg/kg m.c. co 6-8 godzin (nie wolno przekraczać dawki 9 µg/kg m.c.). W premedykacji u dorosłych można stosować 0,3 mg domięśniowo godzinę przed zabiegiem. Jako uzupełniający lek przeciwbólowy u dorosłych podaje się 0,3-0,45 mg dożylnie. Po 10 min od podania domięśniowego stężenie we krwi nie różni się znacznie od stężenia po podaniu dożylnym takich samych dawek leku.
Interakcje
Buprenorfina wiąże się w 96% z białkami osocza: α- i β-globulinami. Metabolizowana jest w wątrobie poprzez CYP3A4 do norbuprenorfiny, która jest słabym agonistą receptora opioidowego μ, i sprzężonych z kwasem glukuronowym metabolitów buprenorfino-3-glukuronidu i norbuprenorfino-glukuronidu. 70% leku wydalane jest w postaci niezmienionej drogą przewodu pokarmowego, pozostała część z moczem. Jednoczesne stosowanie leków hamujących lub indukujących enzym CYP3A4 może teoretycznie odpowiednio zwiększyć lub zmniejszyć skuteczność analgetyczną buprenorfiny, choć nie przeprowadzono badań, by ostatecznie ocenić te interakcje. Ponieważ nie wyjaśniono znaczenia skutków hamowania lub indukowania, nie zaleca się łączenia buprenorfiny z lekami wpływającymi na aktywność izoenzymu CYP3A4.
U pacjentów z niewydolnością wątroby siła i czas działania buprenorfiny mogą ulec zmianie, dlatego pacjenci powinni być poddani dokładnej obserwacji. U chorych z niewydolnością nerek farmakokinetyka buprenorfiny nie zmienia się, dlatego lek może być bezpiecznie stosowany. Ponadto wykazano, że buprenorfina nie jest usuwana podczas hemodializy. W przypadku osób w podeszłym wieku nie jest konieczna modyfikacja dawkowania, gdyż w tej grupie nie obserwowano zmian farmakokinetyki i farmakodynamiki leku.
Przeciwwskazania
Przeciwwskazaniem do podawania buprenorfiny jest nadwrażliwość na substancję czynną, niewydolność oddechowa, leczenie inhibitorami MAO w ciągu 2 tygodni przed zastosowaniem buprenorfiny, męczliwość mięśni, majaczenie alkoholowe, ciąża.
Buprenorfinę należy ostrożnie stosować u pacjentów przyjmujących leki o działaniu hamującym czynność OUN (inne leki opioidowe, leki stosowane w znieczuleniu ogólnym, przeciwhistaminowe, pochodne fenotiazyny, uspokajające, nasenne). W przypadku takiej terapii skojarzonej zaleca się zmniejszenie dawki jednego lub obu leków. Ostrożnie należy podawać pacjentom z zaburzeniami czynności układu oddechowego (np. astmą, przerostem prawej komory serca, zmniejszeniem rezerwy oddechowej, niedotlenieniem narządów, hiperkapnią, uprzednio występującym zahamowaniem oddychania), z urazami głowy, zmianami wewnątrzczaszkowymi oraz innymi zmianami zwiększającymi ciśnienie płynu mózgowo-rdzeniowego, z chorobami utrudniającymi odpływ żółci, z niedoczynnością tarczycy, niewydolnością kory nadnerczy (np. chorobą Addisona), zahamowaniem czynności OUN, psychozą, śpiączką, przerostem gruczołu krokowego lub zwężeniem cewki moczowej, alkoholizmem. Ostrożnie stosować u dzieci. Nie badano bezpieczeństwa podawania buprenorfiny dzieciom <6 m.ż.
W przypadku systemów przezskórnych gorączka i wysoka temperatura otoczenia mogą zwiększyć przenikanie leku przez skórę, teoretycznie stężenie buprenorfiny może się zwiększyć, trzeba więc brać pod uwagę, że nasili się działanie preparatu, a także działania niepożądane.
W przypadku osób uzależnionych od opioidów podanie małych dawek buprenorfiny zapobiegało wystąpieniu zespołu abstynencyjnego, sporadycznie obserwowano euforię.
Działania niepożądane
Terapeutyczne dawki buprenorfiny nie zmniejszają skuteczności standardowych dawek innych leków działających agonistycznie na receptory opioidowe μ. Standardowe dawki innych analgetyków opioidowych można podawać pod koniec działania buprenorfiny i nie ma to wpływu na analgezję. Buprenorfiny nie należy stosować jednocześnie z tzw. lekami Z (zolpidemem i zopiklonem) z uwagi na ryzyko działań niepożądanych ze strony OUN.
Nie ma wystarczających danych, by ocenić bezpieczeństwo podawania buprenorfiny ciężarnym, choć w badaniach na zwierzętach obserwowano toksyczny wpływ na reprodukcję oraz na płód. Ryzyko stosowania u ciężarnych jest nieznane. Buprenorfina przenika do mleka kobiecego w niewielkich ilościach oraz może zmniejszać wydzielanie mleka, dlatego nie należy jej podawać w czasie laktacji.
Buprenorfina wpływa na zdolność prowadzenia pojazdów mechanicznych, zdolność uczestniczenia w ruchu ulicznym i obsługiwania urządzeń mechanicznych w momencie rozpoczęcia leczenia, zmiany dawki i sytuacji, gdy buprenorfina jest podawana z innymi substancjami działającymi ośrodkowo. W przypadku pacjentów przyjmujących ustaloną dawkę preparatu w postaci przezskórnej nie ma konieczności podejmowania restrykcyjnych działań, jeśli nie występują zawroty głowy, senność, zaburzenia widzenia.
Objawy niepożądane występujące bardzo często (≥1/10) obejmują głównie nudności, a w przypadku systemów przezskórnych także rumień i świąd skóry pod plastrem. Częste (≥100 do < 1/10) są wymioty, zaparcia, duszność, zawroty i bóle głowy, obrzęki, zmęczenie, niezbyt często – splątanie, zaburzenia snu, niepokój, senność, niedociśnienie tętnicze, suchość jamy ustnej, osutka. W badaniach porównujących skuteczność i bezpieczeństwo stosowania silnych opioidów u pacjentów z przewlekłym bólem wykazano, że fentanyl częściej powodował nudności, z kolei morfina indukowała więcej zaparć, nudności i wymiotów niż buprenorfina. Pacjenci rzadziej zaprzestają stosowania leku z powodu działań niepożądanych niż chorzy przyjmujący morfinę lub fentanyl.
Ryzyko zespołu abstynencyjnego po odstawieniu buprenorfiny jest mało prawdopodobne, ale po długotrwałym przyjmowaniu leku nie można tego wykluczyć. Ryzyko depresji oddechowej jest mniejsze niż w przypadku stosowania fentanylu, gdyż buprenorfina wykazuje efekt pułapowy dla tego działania niepożądanego (potwierdzone w badaniach eksperymentalnych i na zdrowych ochotnikach). Badania wykazały, że buprenorfina w mniejszym stopniu niż morfina i fentanyl upośledza funkcjonowanie układu odpornościowego, a także w mniejszym stopniu wpływa na funkcję osi podwzgórze-przysadka-gonady i nadnercza, a tym samym ryzyko indukowania zespołu niedoboru androgenów u pacjentów przewlekle stosujących lek jest mniejsze.
Zgodnie z zaleceniami stosowania silnych opioidów u pacjentów w podeszłym wieku, wybierając opioid, należy kierować się farmakokinetyką, farmakodynamiką, ryzykiem działań niepożądanych (preferowane są leki, których farmakokinetyka nie zmienia się w przypadku niewydolności nerek, oraz leki o minimalnym ryzyku rozwoju interakcji lekowych i objawów niepożądanych). W badaniach klinicznych prowadzonych na grupie pacjentów >65 r.ż. nie zaobserwowano zmian skuteczności i bezpieczeństwa buprenorfiny zależnych od naturalnego procesu starzenia się w porównaniu z młodszymi pacjentami.
Ze względu na mechanizm działania buprenorfina jest szczególnie przydatna w leczeniu bólu neuropatycznego, ponieważ nie tylko zmniejsza ból, ale i ogranicza allodynię oraz hiperalgezję. W literaturze można znaleźć kilka doniesień o skuteczności buprenorfiny w zespołach bólu neuropatycznego, jak np. ból po torakotomii, ból fantomowy, neuralgia popółpaścowa, ból korzeniowy, ból centralny. Przezskórne systemy terapeutyczne zawierające buprenorfinę należy zmieniać co 72-96 godzin, należy pamiętać, że w tym zakresie istnieją różnice pomiędzy lekiem oryginalnym i generycznym, a także różnice osobnicze.
Metadon
Mechanizm działania
Metadon jest silnym opioidem stosowanym w uśmierzaniu bólu przewlekłego, głównie u chorych na nowotwór. Jest on syntetycznym lekiem opioidowym o złożonym mechanizmie działania przeciwbólowego. Poza oddziaływaniem na receptor opioidowy μ metadon jest również antagonistą kluczowego dla procesu nocycepcji receptora NMDA (N-metylo-D-asparaginianowego), aczkolwiek istnieją także doniesienia o działaniu tego leku przez receptor δ. Jest on lekiem opioidowym o prawdopodobnie najbardziej złożonej farmakokinetyce. Tylko 1% metadonu utrzymuje się we krwi, pozostała część gromadzi się w tkankach, stąd bardzo długi czas eliminacji leku z organizmu. Metadon jest metabolizowany w wątrobie i ścianie jelit do kilku nieaktywnych metabolitów, a następnie wydalany przez nerki i przewód pokarmowy (nawet do 60% leku). Ilość metadonu wydalanego z moczem zależy od jego kwasowości: spadek pH moczu powoduje zwiększenie wydalania. U chorych z niewydolnością nerek nie zmienia się klirens metadonu, zwiększa się natomiast wydalanie przez przewód pokarmowy (w przypadku anurii jest on niemalże w całości usuwany tą drogą). W związku z tym może być bezpiecznie stosowany u pacjentów w późnym stadium PChN. Niektórzy autorzy zalecają zmniejszenie dawki metadonu o połowę u chorych, u których stężenie kreatyniny przekracza 8 mg/dl. Metadon może być także stosowany przez chorych przewlekle dializowanych. Prawdopodobnie <1% dobowej dawki leku jest usuwane w trakcie hemodializy lub dializy otrzewnowej. Dializa nie wpływa w związku z tym w istotny sposób na stężenie leku we krwi, dzięki czemu możliwe jest utrzymanie stabilnej kontroli bólu. Okres półtrwania metadonu wydłuża się istotnie u chorych z ciężką niewydolnością wątroby, dlatego w tej grupie pacjentów lek należy podawać bardzo rozważnie. Czas eliminacji metadonu jest długi i bardzo zmienny, wynosi 15-60 godzin. Tak więc po zmianie dawkowania stan stacjonarny w surowicy ustala się najwcześniej po upływie kilku dni. Terapię metadonem rozpoczyna się zazwyczaj od dawek 2,5-5 mg/12 h p.o.
Podobnie jak w przypadku innych silnych opioidów ważne są interakcje farmakodynamiczne metadonu z innymi lekami, w tym z benzodiazepinami i innymi lekami działającymi depresyjnie na OUN. Istnieją doniesienia o wystąpieniu zespołu serotoninowego u chorych leczonych metadonem, łącznie z sertraliną, wenlafaksyną i cyprofloksacyną. Metadon może wydłużać odstęp QT, co u pacjentów ze współistniejącymi innymi czynnikami sprzyjającymi występowaniu zaburzeń rytmu serca leczonych dużymi dawkami tego leku i np. haloperydolem, trójpierścieniowymi lekami przeciwdepresyjnymi lub cyprofloksacyną może być przyczyną groźnych dla życia zaburzeń rytmu serca. Z tego względu nie zaleca się również łączenia metadonu z fentanylem.
Metadon charakteryzuje się mniejszym potencjałem uzależniającym, a rozwój zespołu odstawienia w przypadku metadonu trwa dłużej.
Wskazania
Poza terapią bólu przewlekłego metadon jest stosowany w leczeniu uzależnienia od opioidów oraz zespołów abstynencyjnych. W porównaniu z morfiną rzadziej powoduje nudności i wymioty oraz zaparcia. Ze względu na mechanizm działania i antagonizm leku do receptorów NMDA uważa się, że jest on szczególnie przydatny w leczeniu bólu neuropatycznego u chorych na nowotwór. Długi okres półtrwania, możliwość kumulacji w tkankach oraz osobnicze odmienności w farmakokinetyce sprawiają, że dawkowanie metadonu jest zindywidualizowane i powinno być wdrażane przez lekarzy specjalistów leczenia bólu.
Tapentadol
Tapentadol jest pierwszym przedstawicielem nowej klasy analgetyków działających ośrodkowo, o podwójnym mechanizmie działania (MOR-NRI). Ma działanie agonistyczne na receptor opioidowy μ w OUN oraz hamuje wychwyt zwrotny noradrenaliny w zstępującym układzie antynocyceptywnym. Stosunkowo niewielkie powinowactwo tapentadolu do receptora opioidowego μ oraz dodatkowy mechanizm działania – hamowanie wychwytu zwrotnego noradrenaliny – skutkuje zmniejszeniem częstości działań niepożądanych typowych dla agonistów receptora opioidowego μ z zachowaniem jednocześnie silnego działania przeciwbólowego (μ-sparing effect). Tapentadol nie jest prolekiem, jego działanie przeciwbólowe nie jest więc warunkowane aktywacją metaboliczną. Lek nie ma aktywnych metabolitów, a jego stosowanie wiąże się z małym ryzykiem interakcji lekowych (niski poziom wiązania leku do białek osocza, brak wpływu na układ cytochromu P450). W badaniach eksperymentalnych stwierdzono, że wykazuje około 50-krotnie słabsze powinowactwo do receptora opioidowego μ niż morfina, w badaniach klinicznych poprzez silne działanie wzmacniające układ zstępujący noradrenergiczny wywiera efekt analgetyczny (w porównaniu z morfiną tylko 2-3-krotnie słabszy). W badaniach eksperymentalnych i klinicznych stwierdzono dużą skuteczność tapentadolu, zarówno w zespołach bólu ostrego (somatycznego i trzewnego), jak i przewlekłego, w tym bólu neuropatycznego. Ze względu na podwójny mechanizm działania, zwłaszcza hamowanie wychwytu zwrotnego noradrenaliny, tapentadol jest szczególnie skuteczny w leczeniu bólu z komponentem neuropatycznym. W badaniach klinicznych tapentadol stosowany w różnych zespołach bólu przewlekłego wykazywał podobną ogólną skuteczność analgetyczną jak inne silne opioidy (morfina, oksykodon, oksykodon z naloksonem), cechował się jednak statystycznie istotnie mniejszą częstością objawów niepożądanych ze strony przewodu pokarmowego (zaparcia, nudności, wymioty) i ośrodkowego układu nerwowego niż inne silne opioidy. Podczas długoterminowych, rocznych obserwacji nie zaobserwowano rozwoju tolerancji na lek, a po odstawieniu go u 90% pacjentów nie stwierdzono objawów odstawiennych. Ze względu na unikalny mechanizm działania i korzystny profil bezpieczeństwa tapentadol może być korzystnym wyborem w leczeniu wielu zespołów bólowych, w tym bólu przewlekłego z komponentem neuropatycznym. Tapentadol jest lekiem zarejestrowanym w Polsce w postaci tabletek o natychmiastowym (niedostępny na rynku) i przedłużonym uwalnianiu. Wskazaniem do zastosowania tapentadolu o przedłużonym uwalnianiu wg charakterystyki produktu leczniczego (ChPL) jest leczenie bólu przewlekłego o dużym natężeniu u osób dorosłych, który może być właściwie opanowany jedynie po zastosowaniu opioidowych leków przeciwbólowych. Preparat o przedłużonym uwalnianiu dostępny jest w postaci tabletek po 50, 100, 150, 200, 250 mg w opakowaniach po 60 tabletek. Lek jest objęty refundacją w leczeniu przewlekłego bólu o dużym nasileniu w przebiegu chorób nowotworowych – u dorosłych pacjentów, u których nie uzyskano odpowiedniej kontroli bólu po zastosowaniu morfiny o zmodyfikowanym lub przedłużonym uwalnianiu lub jej nietolerancji.
Oksykodon
Mechanizm działania
Oksykodon jest półsyntetycznym opioidem uzyskiwanym z alkaloidu opium – tebainy. Jest on agonistą receptora μ i κ o sile działania przeciwbólowego porównywalnej z morfiną. Prawdopodobnie aktywność oksykodonu w stosunku do receptorów opioidowych κ w obszarze trzewnym może wyjaśniać jego lepszą od innych opioidów skuteczność w leczeniu bólu trzewnego. W porównaniu z morfiną oksykodon cechuje wysoka biodostępność po podaniu p.o. (42-87% oksykodon vs. 22-48% morfina). Oksykodon jest metabolizowany przy udziale CYP3A4 do noroksykodonu oraz przy udziale CYP2D6 do oksymorfonu. Oksymorfon cechuje 14-krotnie większe w porównaniu z oksykodonem powinowactwo do receptora opioidowego μ, ale ze względu na niewielkie (ok. 2%) stężenie jego udział w całkowitym efekcie analgetycznym wydaje się znikomy. Udział w metabolizmie oksykodonu dwóch typów cytochromu P450: CYP3A4 i CYP2D6, sprawia, że ryzyko interakcji farmakokinetycznych z innymi lekami jest niewielkie. Niezmieniony oksykodon oraz jego metabolity są wydalane głównie przez nerki. W niewydolności nerek stężenie oksykodonu zwiększa się, a jego okres półtrwania istotnie wydłuża – w niektórych przypadkach obserwowano wydłużenie okresu półtrwania nawet do 26 godzin. Podobnie u chorych z upośledzoną czynnością wątroby obserwuje się wzrost stężenia oraz wydłużenie okresu półtrwania (nawet do 24 godzin).
Dawkowanie
W przypadku oksykodonu nie obserwuje się efektu pułapowego, zależność dawka-efekt przeciwbólowy ma charakter liniowy. Może on natomiast powodować działania niepożądane, podobnie jak inne opioidy, takie jak sedacja, nudności, wymioty, zaparcia, depresja oddechowa. Stosowany jest drogą doustną lub dożylną, przy czym współczynnik konwersji przy zamianie oksykodonu podawanego drogą dożylną na doustną wynosi 1:2. W przypadku zamiany morfiny na oksykodon współczynnik konwersji wg Palliative Care Formulary 3 wynosi 1,5:1 (tzn. zamiana z 15 mg/24 h morfiny na 10 mg/24 h oksykodonu), wg producenta 2:1 (tzn. zamiana 20 mg/24 h morfiny na 10 mg/24 h oksykodonu). Warto jednak pamiętać, że z powodu profilu receptorowego działania oksykodonu dawki równoważne powinny być przeliczane bardzo ostrożnie. Najbezpieczniejszą metodą ustalenia dawki analgetycznej jest miareczkowanie. Preparaty doustne oksykodonu o kontrolowanym uwalnianiu charakteryzują się innowacyjnym dwufazowym modelem uwalniania i wchłaniania umożliwiającym uzyskanie szybkiego (w ciągu godziny) początku analgezji trwającej przez 12 godzin. Tabletki oksykodonu o kontrolowanym uwalnianiu dostępne są w dawkach 5 mg, 10 mg, 20 mg, 40 mg i 80 mg. Na rynku jest dostępny i refundowany w wybranych wskazaniach lek będący połączeniem oksykodonu z antagonistą receptora opioidowego – naloksonem w preparacie o kontrolowanym uwalnianiu. Preparat ten jest stosowany zarówno u pacjentów chorych na nowotwór, jak i w leczeniu bólu nienowotworowego. Oksykodon zapewnia analgezję dzięki dużej biodostępności po podaniu doustnym, a nalokson przeciwdziała zaburzeniom defekacji wywołanym opioidami, blokując połączenie oksykodonu z receptorami opioidowymi w ścianie jelit. Nalokson o kontrolowanym uwalnianiu nie wykazuje natomiast działania ośrodkowego i dzięki temu nie odwraca efektu analgetycznego oksykodonu. Jest to możliwe dzięki bardzo intensywnemu metabolizmowi wątrobowemu naloksonu (w 97% ulega efektowi pierwszego przejścia przez wątrobę), który zabezpiecza przed przedostawaniem się większych ilości naloksonu do krążenia systemowego i w następstwie przed osłabieniem efektu analgetycznego. W badaniach ustalono, że optymalne proporcje oksykodonu do naloksonu powinny wynosić 2:1. Ważne dla skuteczności preparatu są wydolność wątroby, która zapewnia odpowiednio intensywny metabolizm naloksonu, oraz system kontrolowanego uwalniania naloksonu, który zapobiega efektowi wysycenia enzymów wątrobowych przez nalokson. Oksykodon z naloksonem cechuje się dobrą skutecznością przeciwbólową podobną do samego oksykodonu. Oksykodon jest opioidem z wyboru w leczeniu bólu trzewnego.
Petydyna
Petydyna należy do analgetyków opioidowych, jest 10-krotnie słabsza od morfiny i ma działanie antycholinergiczne, dlatego przez wiele lat funkcjonował mit, że jest jedynym opioidem, który można stosować w bólach kolkowych. Metabolit petydyny – norpetydyna – zwiększa jednak ryzyko przetrwałego bólu pooperacyjnego, gdyż powoduje uwalnianie w strukturach OUN licznych czynników pronocyceptywnych oraz działa cholinolitycznie (upośledza funkcjonowanie endogennych szlaków kontroli bólu). W żadnym wypadku nie może być stosowana w położnictwie, ponieważ kumuluje się w kobiecym mleku i działa neurotoksycznie na mózg noworodka karmionego piersią. Może spowodować u noworodka ciężkie objawy niepożądane ze zgonem włącznie. Petydyna z uwagi na aktualną wiedzę nie może być rekomendowana w leczeniu bólu.
Opioidowe leki przeciwbólowe u ciężarnej (kategoria C wg FDA)
Leki te można stosować u kobiet w ciąży i karmiących tylko wówczas, gdy korzyści z ich podania przewyższają ryzyko niekorzystnego wpływu na płód. Jakkolwiek stosowanie opioidów w tym czasie staje się istotnym problemem (np. w USA 20% ciężarnych zażywa opioidowe leki przeciwbólowe, a ok. 4,4% ciężarnych jest uzależnionych od opioidów). Opioidy szczególnie w przypadku dłuższego stosowania mogą powodować:
- gorszy rozwój fizyczny, poznawczy dziecka (tylko dzieci kobiet, które w ciąży były uzależnione od opioidów i innych substancji)
- objawy odstawienia (30-90% noworodków)
- depresję oddechową noworodka (zwłaszcza w przypadku stosowania kodeiny i dihydrokodeiny).
Uważa się, że opioidy nie działają teratogennie, jakkolwiek pojawiają się publikacje opisujące przypadki wad rozwojowych u dzieci po stosowaniu przez ciężarne:
- tramadolu (wady przegrody międzyprzedsionkowej lub międzykomorowej, stopa końsko-szpotawa)
- kodeiny (ryzyko wytrzewienia jelit, rozszczepu kręgosłupa)
- kodeiny, dihydrokodeiny (wady przegrody międzyprzedsionkowej lub międzykomorowej, wady odpływu lewokomorowego, zespół hipoplazji lewego serca)
- oksykodonu (stenozy zastawki płucnej)
- metadonu (podwyższone ryzyko zeza u dzieci, zmniejszenie obwodu głowy i długości ciała, niższa masa urodzeniowa).
Nalbufina jest opioidowym lekiem przeciwbólowym o korzystnym profilu bezpieczeństwa dla kobiet w ciąży (kategoria B wg FDA), ponieważ jest antagonistą receptora opioidowego MOR (μ), co zmniejsza istotnie ryzyko działań niepożądanych charakterystycznych dla innych opioidów.
Leki opioidowe należy stosować tylko, gdy wskazania nie budzą wątpliwości i nie ma innej alternatywy:
- długoterminowe stosowanie opioidów u ciężarnych jest dopuszczalne w przypadku terapii substytucyjnej oraz w szczególnych sytuacjach, kiedy natężenie bólu uzasadnia wybór tej grupy leków
- gwałtowne odstawienie opioidów w ciąży nie jest zalecane – może spowodować obumarcie płodu lub przedwczesny poród
- silnie działające opioidy można odstawiać stopniowo, tak by uniknąć zespołu odstawiennego u matki i płodu
- u noworodków może wystąpić depresja oddechowa lub zespół odstawienny
- depresja oddechowa u noworodka może się pojawić w przypadku podania opioidu krótko przed porodem, np. w analgezji porodu; może być potrzebne podanie antagonisty
- zespół odstawienny u noworodka może się pojawić w ciągu dni lub tygodni po porodzie, może mieć różne nasilenie
- w leczeniu zespołu odstawiennego wystarczy karmienie noworodka piersią
- w przypadku dużego nasilenia objawów potrzebna jest substytucja opioidem.
Bezpieczeństwo stosowania analgetyków opioidowych
Podczas stosowania analgetyków opioidowych mogą występować działania niepożądane związane z powinowactwem tych leków do receptorów opioidowych lub efektami farmakologicznymi tej grupy leków.
Do najczęstszych działań niepożądanych obserwowanych podczas stosowania analgetyków opioidowych należą:
- sedacja, splątanie, jakościowe zaburzenia zachowania (szczególnie w populacji geriatrycznej)
- nudności i wymioty
- zaburzenia defekacji (OIBD – opioid-induced bowel dysfunction) – nie należy ich utożsamiać z zaparciem z uwagi na patomechanizm
- świąd skóry
- działanie immunosupresyjne
- depresja oddechowa, która przy prawidłowo monitorowanej terapii jest powikłaniem niezmiernie rzadkim.
Większość objawów niepożądanych w miarę trwania terapii słabnie, z wyjątkiem zaburzeń funkcji przewodu pokarmowego, co jest ściśle skorelowane z obecnością receptorów opioidowych w przewodzie pokarmowym.
Zlecając pacjentowi analgetyki opioidowe, należy się liczyć z tym, że wystąpią objawy niepożądane ze strony przewodu pokarmowego, szczególnie nudności, wymioty, bóle brzucha oraz OIBD.
W przypadku nudności i wymiotów indukowanych przez analgetyki opioidowe można stosować leki z grupy antagonistów receptora H1 wykazujące efekt ośrodkowy (dimenhydrynat; pochodne fenotiazyny – lewomepromazynę, tietylperazynę), hydroksyzynę, inne leki cholinolityczne, metoklopramid, setrony (ondansetron, granisetron) oraz deksametazon. Metoklopramidu nie należy stosować, gdy analgetyk opioidowy jest metabolizowany przez CYP2D6 (szczególnie tramadol), ponieważ jest on silnym inhibitorem izoenzymu CYP2D6. Podobnie podczas terapii tramadolem nie można stosować setronów, które antagonizują efekt analgetyczny leku związany z wpływem na układ serotoninergiczny. W przypadku wymiotów o znacznym nasileniu skuteczni są antagoniści receptorów neurokininowych, np. aprepitant, netupitant.
Według różnych statystyk aż 70-100% pacjentów przyjmujących analgetyki opioidowe ma zaburzenia defekacji, które z jednej strony znacznie upośledzają komfort życia, z drugiej niejednokrotnie uniemożliwią osiągnięcie skutecznej dawki analgetycznej i pogarszają przestrzeganie zaleceń przez pacjenta. Pacjenci leczeni opioidami ze znacznym wysiłkiem i zbyt rzadko oddają twardy stolec, czemu towarzyszy poczucie niepełnego wypróżnienia, czasem przestają się wypróżniać.
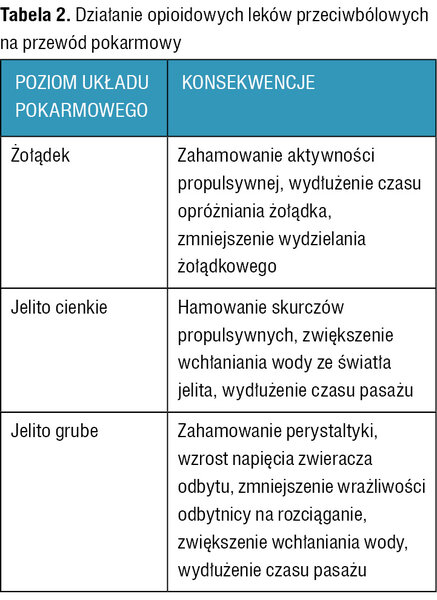
Tabela 2. Działanie opioidowych leków przeciwbólowych na przewód pokarmowy
Zaburzenia defekacji są najbardziej dolegliwym objawem niepożądanym analgetyków opioidowych, na które nie obserwuje się tolerancji. Do grupy ryzyka tego powikłania należą pacjenci leczeni opioidami: w podeszłym wieku, przyjmujący wiele leków, które zaburzają perystaltykę przewodu pokarmowego, oraz pacjenci w terminalnym okresie choroby nowotworowej.
Perystaltyka przewodu pokarmowego jest regulowana przez ośrodkowy układ nerwowy, układ nerwowy jelita, a także wydzielane do przewodu pokarmowego hormony. Opioidowe leki przeciwbólowe, oddziałując na receptory opioidowe typu μ i κ zlokalizowane w mięśniówce, splotach mięśniówki, w splocie podśluzówkowym jelita cienkiego i grubego oraz modulując funkcje receptorów opioidowych na poziomie rdzenia kręgowego, spowalniają perystaltykę i wpływają na funkcje wydzielnicze przewodu pokarmowego, co dodatkowo nasila zaburzenia perystaltyki (tab. 2).
Ponadto opioidy hamują wydzielanie acetylocholiny ze splotu Auerbacha, powodują relaksację mięśniówki gładkiej, zmniejszają wydzielanie trzustkowe oraz zmniejszają wydzielanie żółci, co dodatkowo nasila zaburzenia perystaltyki przewodu pokarmowego.
W przypadku zaparć indukowanych przez opioidy leczenie objawowe jest mało skuteczne, a zmiana stylu życia oraz zmiana diety często niemożliwe. OIBD po opioidach występuje częściej u pacjentów, którzy są lub byli leczeni winkrystyną, winblastyną, pochodnymi platyny, taksanami oraz talidomidem. Mogą się nasilać wskutek równoczesnego podawania niesteroidowych leków przeciwzapalnych, trójpierścieniowych leków przeciwdepresyjnych, leków antycholinergicznych, leków przeciwdrgawkowych wykorzystywanych jako koanalgetyki, a także setronów stosowanych w objawowym leczeniu nudności i wymiotów.
W przypadku rozpoczynania terapii opioidami zaleca się wdrożenie profilaktyki przeciw zaburzeniom defekacji, ale im dłużej trwa leczenie opioidami, tym mniej skuteczne stają się stosowane długotrwale leki przeczyszczające. Co więcej, same leki przeczyszczające mogą indukować objawy niepożądane – bóle, wzdęcia oraz zaburzenia perystaltyki przewodu pokarmowego nasilające zaburzenia defekacji (jako efekt z odbicia).
Najskuteczniejszą metodą prewencji i zmniejszenia nasilenia OIBD jest podawanie opioidów z naloksonem (antagonistą receptorów opioidowych).
Inne leki, które można stosować do leczenia zaburzeń defekacji indukowanych przez opioidy, to bromek metylonaltreksonu, naloksegol oraz alwimopan.
Antagoniści receptorów opioidowych
Nalokson
To pochodna oksymorfanu. Jest kompetycyjnym, nieselektywnym antagonistą wszystkich trzech receptorów opioidowych. Znosi działanie przeciwbólowe, depresyjny wpływ opioidów na ośrodek oddechowy oraz ich działanie sedatywne. Jest podawany pozajelitowo (nie wchłania się z przewodu pokarmowego) lub doustnie w połączeniu z oksykodonem w preparacie o kontrolowanym uwalnianiu. Jest także stosowany w leczeniu uzależnienia od alkoholu.
Naltrekson
Ma podobny profil selektywności receptorowej jak nalokson, ale działa kilka razy silniej oraz kilkakrotnie dłużej niż nalokson i może być stosowany doustnie. Tak jak nalokson znalazł zastosowanie w leczeniu uzależnień alkoholowych.
Bromek metylonaltreksonu
Jest selektywnym antagonistą receptora opioidowego μ stosowanym w leczeniu zaparć wywołanych opioidami u pacjentów, u których odpowiedź na środki przeczyszczające jest niewystarczająca. Ze względu na to, że metylonaltrekson nie przechodzi przez barierę krew-mózg, nie obserwuje się osłabienia działania przeciwbólowego ani objawów odstawiennych po jego zastosowaniu.
Podawany jest we wstrzyknięciach podskórnych w pojedynczej dawce w zależności od masy ciała pacjenta: 8 mg (przy masie ciała ≤61 kg) lub 12 mg (>61 kg) co 2 dni lub w dłuższych odstępach, w zależności od potrzeby. Do 4 godzin od podania leku wypróżnia się 50-60% pacjentów. W przypadku braku efektu dawkę można powtórzyć po 24-48 godzinach.
Metylonaltrekson może powodować objawy niepożądane: bóle brzucha, wzdęcia, nudności, rzadziej zawroty głowy i biegunkę, które zazwyczaj mają łagodny lub umiarkowany charakter i są zwykle związane z defekacją. Jest przeciwwskazany w przypadku podejrzewanej lub potwierdzonej niedrożności przewodu pokarmowego lub ostrego brzucha wymagającego interwencji chirurgicznej oraz ciężkiej niewydolności wątroby i nerek.
Naloksegol
To antagonista obwodowych receptorów opioidowych μ. Jest pochodną naloksonu, ale w porównaniu z nim ma mniejszą zdolność przenikania do mózgu, dzięki czemu blokuje receptory opioidowe μ w przewodzie pokarmowym, nie oddziałując na receptory znajdujące się w mózgu.
Abstract
Opioid analgesics
Opioids are one of the main classes of drugs used for pain management. Experience demonstrates that rational pain pharmacotherapy based on a sound understanding of the mechanism of drug action, pharmacokinetics, adverse reactions and interactions with different types of medicines helps to effectively relieve pain in most cases, i.e. in as many as 85-95% of patients. With adequate analgesics and a suitable dosing regimen, it is possible to ensure earlier patient mobilization, to reduce the frequency of complications, and to decrease the risk of chronic post-surgical pain.
- 1. Malec-Milewska M, Woroń J (red.). Kompendium leczenia bólu. Warszawa: Medical Education, 2017.
- 2. Bell RF. Cancer pain: analgesics and co-analgesics. W: Evidence-Based Chronic Pain Management (ed. Stannard CF. i wsp.). Chichester: BMJ Books, 2010.
- 3. Bennett MI. Treatment of cancer pain. W: Pain 2012. Refresher courses 14th World Congress on Pain. (ed. Tracey I.). Seattle: IASP Press, 2012. 4. Caraceni A, Hanks G, Kaasa S, et al. Use of opioid analgesics in the treatment of cancer pain: evidence-based recommendations from the EAPC. Lancet Oncol 2012; 13: 58-68.
- 5. Current Topics in Pain: 12th World Congress on Pain (ed. Castro-Lopez J.). Seattle: IASP Press, 2009.
- 6. Dean M. Opioids in renal failure and dialysis patients. J Pain Sym Manage 2004;28:497-504.
- 7. Wells BG, DiPiro JT, Schwinghammer TL, et al. Pharmacotherapy Handbook. New York: McGrawHill, 2016.
- 8. Hutchison LC, Sleeper RB. Fundamentals of Geriatric Pharmacotherapy. Bethesda: ASHP Publications, 2015.
- 9. Bazire S. Psychotropic Drug Directory 2014. Dorsington: Lloyd-Reinhold Communications, 2014.
- 10. Hochadel MA. Mosby’s Drug Reference for Health Proffesions. St Louis: Elsevier, 2016.
- 11. Preston CL. Stockley’s Drug Interactions 2015. London: Pharmaceutical Press, 2014.
- 12. Hansten PD, Horn JR. Top 100 Drug Interactions 2017. Freeland: H&H Publications, 2015.
Pierwszy artykuł:

 Dodaj do ulubionych
Dodaj do ulubionych
