Jawną klinicznie encefalopatię wątrobową ze względu na czas trwania objawów dzieli się na: epizodyczną, nawracającą i przewlekłą (ryc. 1). Przewlekła EW występuje stale, lecz jej nasilenie się zmienia. Epizodyczną postać rozpoznaje się w przypadku nawrotu objawów encefalopatii wątrobowej po okresie dłuższym niż 6 miesięcy, a nawracającą postać, jeśli epizody EW powtarzają się w krótszych odstępach czasu. Przyczyną nawracającej EW są najczęściej czynniki indukujące.
Czynniki indukujące
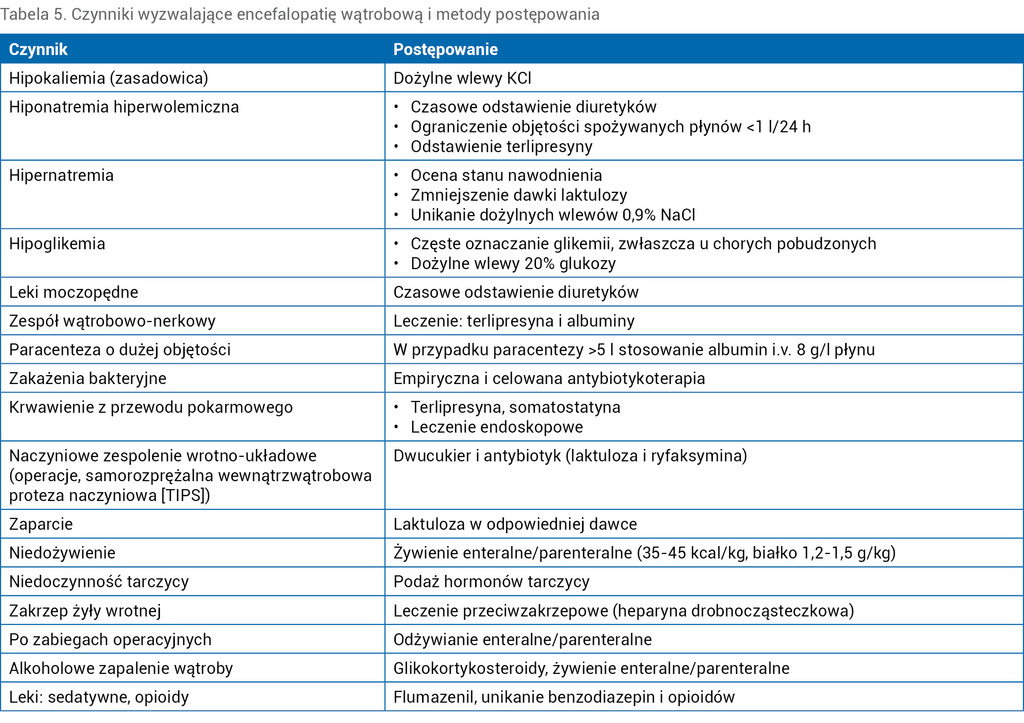
Tabela 5. Czynniki wyzwalające encefalopatię wątrobową i metody postępowania
Postać indukowana encefalopatii wątrobowej stanowi około 90% wszystkich przypadków. Czynniki indukujące rozwój tej patologii mogą być pochodzenia endogennego lub jatrogennego. Listę czynników odpowiedzialnych za pojawienie się EW przedstawiono w tabeli 5. Z patogenetycznego punktu widzenia indukcja encefalopatii wątrobowej polega na promocji jednego lub kilku mechanizmów odpowiedzialnych za rozwój tego zaburzenia. W przypadku stwierdzenia czynnika indukującego należy przystąpić do szybkiego jego wyeliminowania. Najczęstszymi czynnikami są: infekcje bakteryjne, duże dawki diuretyków, zaparcie bądź zaburzenia elektrolitowe.
Diagnostyka różnicowa
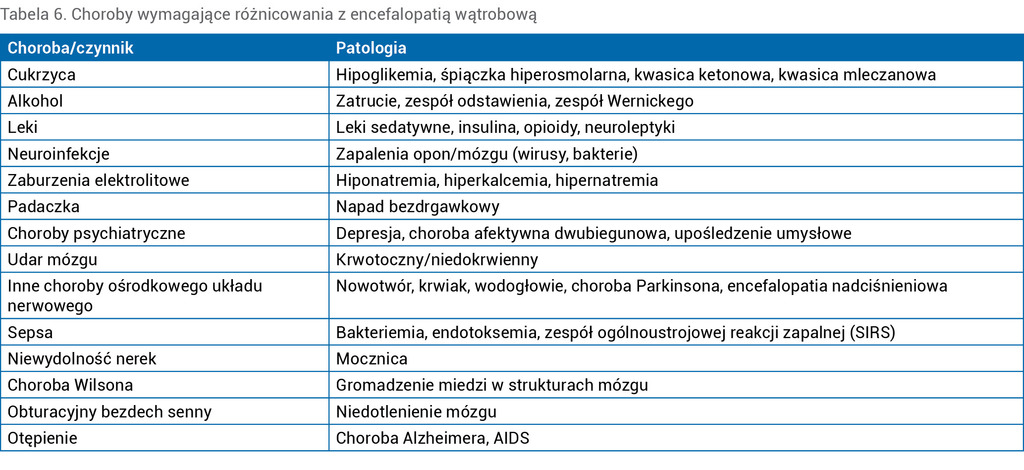
Tabela 6. Choroby wymagające różnicowania z encefalopatią wątrobową
W diagnostyce różnicowej EW należy wziąć pod uwagę zaburzenia neurologiczne związane z otępieniem starczym, alkoholizmem, chorobą Wilsona, hiponatremią, encefalopatią pourazową, infekcyjną i nadciśnieniową (tab. 6). Często w celu wykluczenia innych chorób OUN konieczne jest wykonanie badania obrazowego mózgu. Obecność zmian zwyrodnieniowych mózgu spowodowanych chorobami naczyniowymi, a także zespół Wernickego stanowią przeciwwskazanie do transplantacji wątroby.
U chorych pobudzonych należy wykluczyć hipoglikemię i zespół odstawienia alkoholowego. W leczeniu osób pobudzonych w stopniu utrudniającym opiekę lekarską i pielęgniarską preferuje się haloperydol, a spośród benzodiazepin można rozważyć lorazepam, którego okres półtrwania jest znacznie krótszy od diazepamu.
Hiponatremia
Hiponatremia u chorych z niewyrównaną marskością wątroby jest związana z nadmierną objętością wody, która rozcieńcza sód. Jest to najczęstsze zaburzenie elektrolitowe u chorych z marskością wątroby i jednocześnie oznaka zaawansowania marskości brana pod uwagę w ocenie pilności przeszczepienia tego narządu. Z tego powodu stężenie osoczowe sodu zostało włączone do skali punktowej MELD (MELD-Na) oceniającej krótkoterminową prognozę przeżywalności.
Przewlekłą hiponatremię definiuje się jako spadek stężenia sodu <136 mmol/l, do którego doszło w ciągu kilku tygodni. Stężenie sodu <120 mmol/l określa się mianem ciężkiej hiponatremii. Objawy przewlekłej hiponatremii z powodu adaptacji mózgowia mogą być dyskretne, dlatego rzadko są rozpoznawane, a jeżeli zostają zauważone, to są przypisywane encefalopatii wątrobowej. W rzeczywistości hiponatremia może nasilać lub wyzwalać EW. Do objawów przewlekłej hiponatremii należą: zaburzenia koncentracji uwagi, utrata łaknienia, nudności, bóle głowy, upośledzenie funkcji poznawczych oraz zaburzenia równowagi i chodu z tendencją do upadków. Objawy te istotnie upośledzają jakość życia pacjenta i pogarszają przebieg kliniczny marskości wątroby. W okresie ciężkiej hiponatremii pojawiają się głębokie zaburzenia świadomości z drgawkami. Należy je różnicować z drgawkami, które mogą być objawem obrzęku mózgu w ONW.
Postępowanie u chorego z hiponatremią hiperwolemiczną w przypadku stężeń sodu <125 mmol/l polega na odstawieniu diuretyku, ograniczeniu w diecie zawartości sodu do 80 mmol/24 h i podaży płynów oraz rozważeniu przyspieszonej kwalifikacji do przeszczepienia wątroby. Objętość wypijanych płynów nie powinna przekraczać 1 l/24 h (spożywanie płynów tylko w okresie pragnienia). W przypadku stężenia sodu <120 mmol/l można zastosować 0,9% roztwór NaCl w połączeniu z furosemidem lub torasemidem, najlepiej w ciągłym wlewie dożylnym.
Patogeneza encefalopatii wątrobowej
Rola amoniaku
Amoniak jest wytwarzany w jelitach w przebiegu bakteryjnego metabolizmu amin, aminokwasów, puryn i mocznika. Za produkcję amoniaku odpowiadają głównie bakterie Gram(–) z gatunku Enterobacteriaceae i beztlenowce, bakterie te są bowiem wyposażone w enzymy – ureazę i proteazy. W mniejszych ilościach amoniak jest wytwarzany w nabłonku jelitowym. W warunkach fizjologicznych amoniak, który przedostał się do krwiobiegu, jest metabolizowany głównie w wątrobie, a śladowo w mięśniach szkieletowych i mózgu. W hepatocytach strefy obwodowej płacika wątrobowego amoniak jest przekształcany do mocznika (cykl Krebsa), a w hepatocytach strefy centralnej płacika – do glutaminy. Obydwa produkty są eliminowane z ustroju drogą nerkową.
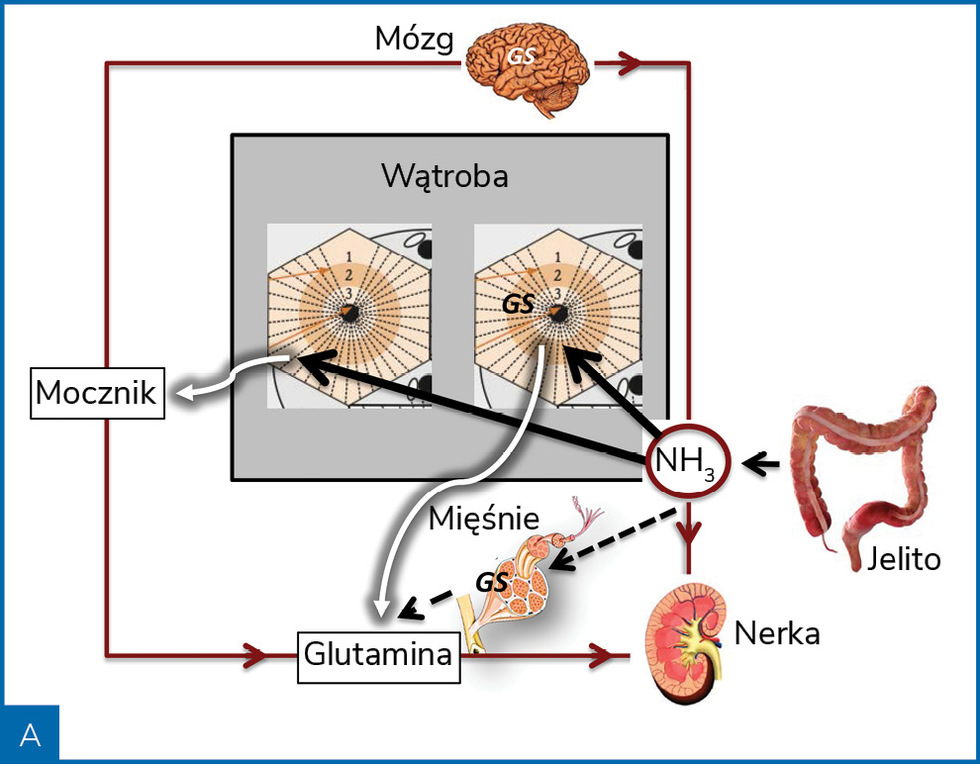
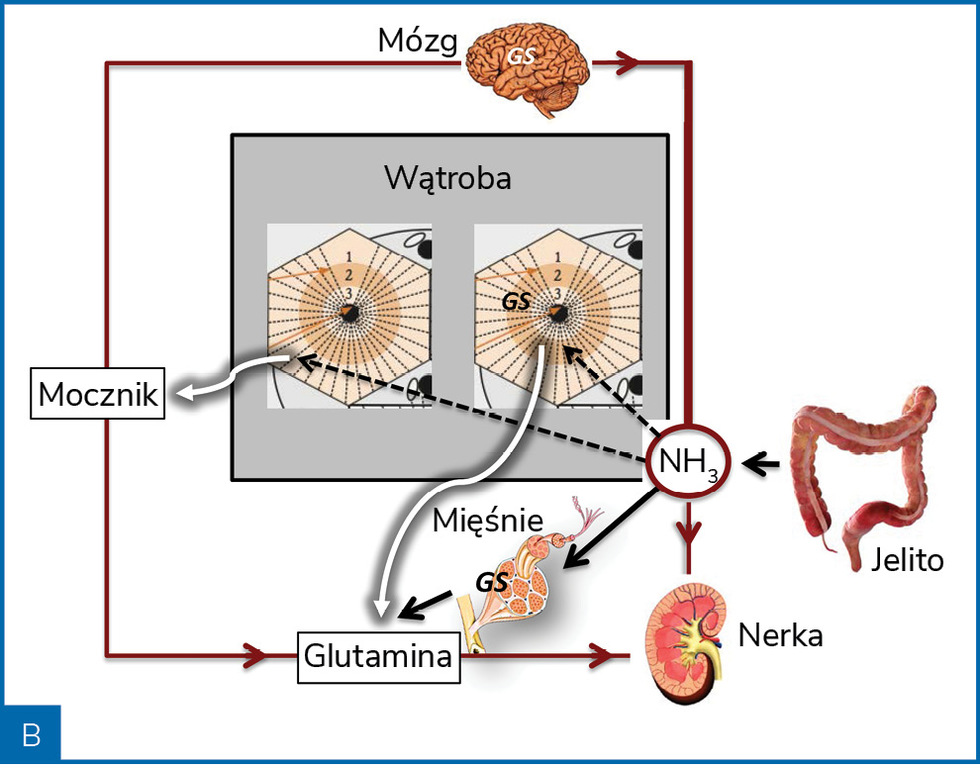
Rycina 2. Poglądowa rycina przedstawiająca metabolizm amoniaku (NH3) w warunkach zdrowia (A) i marskości wątroby (B)
W niewyrównanej marskości wątroby wydolność metaboliczna wątroby w zakresie eliminacji amoniaku zmniejsza się o około 80% i rośnie wtedy udział w jego metabolizmie mięśni i mózgu (ryc. 2). Rola tkanki mięśniowej w eliminacji amoniaku jest jednak ograniczana przez sarkopenię, często obecną w późnej marskości wątroby. Wysokie stężenia amoniaku uszkadzają włókna mięśniowe, co stwarza mechanizm błędnego koła5.
Stężenia amoniaku we krwi chorych z encefalopatią wątrobową są 2-5-krotnie podwyższone, a dodatkowe wzrosty są indukowane przez bogatobiałkowy posiłek lub wysiłek fizyczny, a także krwawienie z przewodu pokarmowego i zaburzenia w składzie ilościowym lub jakościowym mikrobioty jelitowej (dysbioza). U chorych z dyskretnymi objawami EW większą wartość diagnostyczną ma oznaczenie stężenia amoniaku po posiłku. Stężenie amoniaku może być wartościowym badaniem w monitorowaniu wyników leczenia EW. Pomiar stężenia amoniaku metodą enzymatyczną jest badaniem wymagającym, ponieważ amoniak jako substancja gazowa ulatnia się z próbki osocza, a więc czas od pobrania do oznaczenia powinien być możliwie krótki. Pobieranie krwi tętniczej i chłodzenie próbki poprawia wiarygodność oznaczeń. Ostatnio wprowadzono przyłóżkowe pomiary stężenia amoniaku oparte na metodzie spektrofotometrycznej.
Amoniak jako substancja gazowa przenika przez barierę krew–mózg, gdzie hamuje komórkowe potencjały błonowe oraz osłabia czynność neurotransmiterów. Jednakże stężenie amoniaku nie koreluje z kliniczną ekspresją encefalopatii wątrobowej, co wskazuje na współudział innych toksycznych dla mózgu produktów bakteryjnych, takich jak: merkaptany, kwas γ-aminomasłowy, fenole, krótko- i średniołańcuchowe kwasy tłuszczowe oraz oksyndole. Te ostatnie, hamując czynność kanałów sodowych w mózgu, mogą prowadzić do zaburzeń świadomości i śpiączki.
Rola ogólnoustrojowego zespołu zapalnego
Ważnym elementem patogenetycznym EW jest ogólnoustrojowa reakcja zapalna (SIRS – systemic inflammatory response syndrome), która w zakresie OUN może wyczerpywać zasoby energetyczne, zmieniać wielkość przepływu krwi i funkcjonowanie bariery krew–mózg. Ogólnoustrojowa reakcja zapalna w niewyrównanej marskości wątroby jest indukowana przez endotoksemię. Jej nasilenie zależy od wielkości nadciśnienia wrotnego i stopnia niewydolności wątroby. Główną przyczyną endotoksemii jest utrata szczelności bariery jelitowej w warunkach nadciśnienia wrotnego. Znaczenie patogenetyczne mają też inne czynniki, jak zespół rozrostu bakteryjnego spowodowany zmniejszeniem aktywności motorycznej jelita, wzrostem pH żołądka, a nierzadko stosowaniem leków z grupy inhibitorów pompy protonowej, a także upośledzeniem funkcjonowania układu immunologicznego w marskości wątroby. Rozrost flory bakteryjnej jelita cienkiego (SIBO – small intestinal bacterial overgrowth) występuje aż u 70% chorych z zaawansowaną marskością wątroby (klasa B i C według skali Childa-Pugha). Ponadto SIBO odpowiada za wydłużenie czasu tranzytu jelitowego.